
INTRODUCTION

The immunobullous diseases are characterised by cutaneous and mucosal blisters due to pathogenic antibodies which target structural antigens in the skin.
The keratinocytes of the epidermis are held together by a family of proteins called the desmosomes.
In superficial blistering diseases (eg pemphigus vulgaris) these proteins can be targeted and you get superficial blistering
The most clinically relevant tends to be the transmembrane proteins desmoglein 1 and 3
In deeper blistering disorders (eg pemphigoid disorders), the split involves the basement membrane zone:
The inferior part of basal keratinocytes
The basement membrane (lamina lucida and lamina densa)
The upper dermis
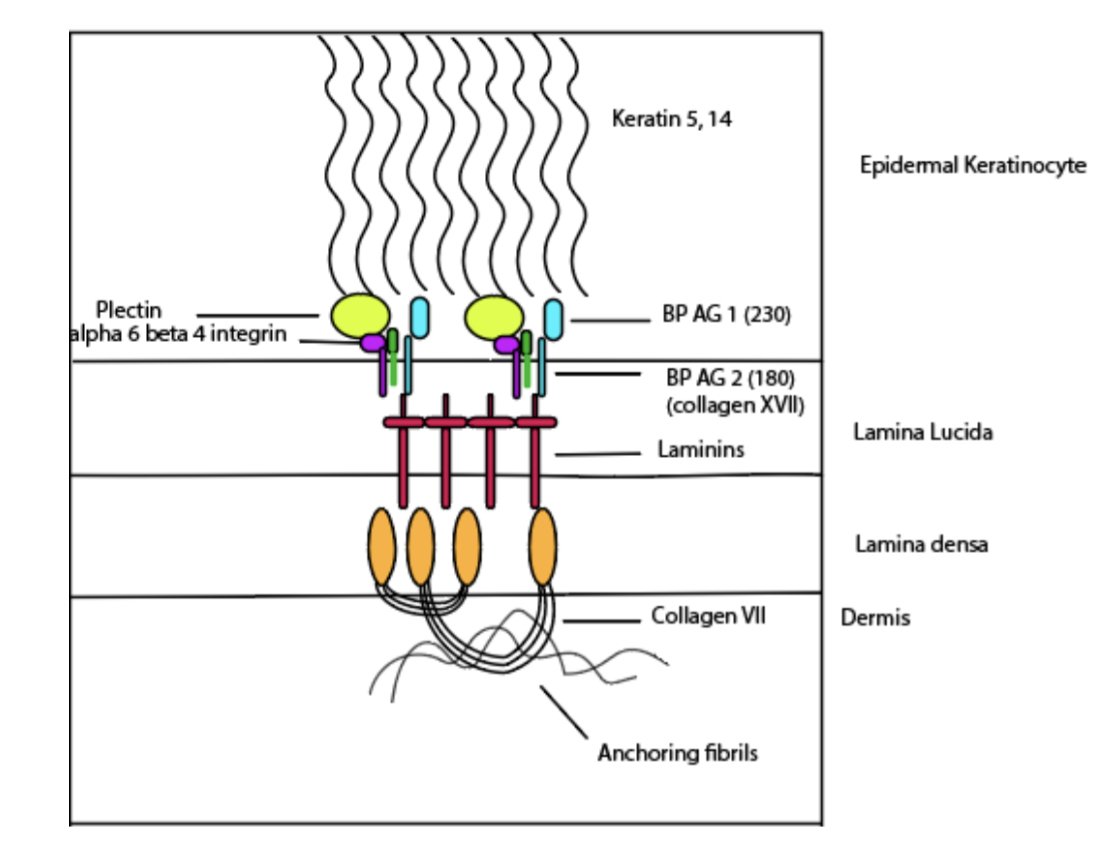
The hemidesmosomes are the collection of proteins involved in keeping the basal keratinocytes attached to the extracellular matrix in the dermis

The clinical picture depends on the depth of blistering:
High in the epidermis (pemphigus diseases): shallow blisters that often present as erosions rather than intact blisters
Around the dermo-epidermal junction (pemphigoid diseases): much more tense blisters
At or below the lamina densa: tense blistering that often results in scarring
DIFFERENTIAL DIAGNOSIS BLISTERING RASH
Blistering Disorders
Differential Diagnosis Overview
- Pemphigus vulgaris
- Pemphigus foliaceus
- Paraneoplastic pemphigus
- IgA pemphigus
- Bullous pemphigoid
- Mucous membrane pemphigoid
- Pemphigoid gestationis
- Dermatitis herpetiformis
- Linear IgA disease
- Epidermolysis bullosa acquisita
- Epidermolysis bullosa (not acquired form)
- HSV
- Herpes zoster
- Hand-foot-mouth disease
- Bullous impetigo
- Staphylococcal scalded skin syndrome
- Severe cellulitis/necrotising fasciitis
- Bullous tinea
- Bullous scabies
- Contact dermatitis (e.g., severe hair dye allergy)
- Phyto-photo dermatitis (plant dermatitis)
- Vesicular hand dermatitis
- Bullous lupus
- Severe vasculitis
- Oedema blisters (e.g., gross oedema in legs)
- Trauma - burns, friction, scalds
- Insect bites
DIAGNOSTIC APPROACH
A standardised approach should include:
Clinical assessment: History, examination, disease activity assessment
Biopsy for H&E (lesional skin)
Biopsy for direct immunofluorescence (peri-lesional skin, within 1cm)
+/- Blood tests: Indirect immunofluorescence +/- ELISA
SKIN BIOPSY
For H&E:
Take a 4mm punch biopsy from lesional area containing a bit of blister and surrounding skin
Send in standard formalin pot
For Direct Immunofluorescence (DIF):
Take a 4mm punch from peri-lesional normal skin (within 1cm of active blister)
Preferably from non-inflamed skin
Send in Michel's medium
HISTOLOGY (H&E)
H&E by itself can’t give you a full diagnosis but it can give you some clues to the diagnosis based on:
Level of split:
Subcorneal
Intraepidermal
Subepidermal
Nature of inflammatory infiltrate:
Neutrophils
Eosinophils
Lymphocytes
Cell-poor
DIRECT IMMUNOFLOURESCENCE (DIF)
DIF is the gold-standard diagnostic tool for immunobullous disease
It allows us to visualise the immune system attacking the skin by making the autoantibodies glow under a microscope
This is performed on tissue from a biopsy
Principle:
Sample: Performed on a biopsy of perilesional skin
Mechanism: Laboratory antibodies "tagged" with fluorescent dye are added to the tissue.
Binding: These lab antibodies seek out and idenify antibodies/proteins (IgG, IgM, IgA, C3 +/- fibrinogen) which is bound to the tissues
Result: Under a fluorescent microscope, the "tagged" areas glow a bright green and we can see the patterns of where and how they are deposited
Result:
DIF is used to narrow down the diagnosis according to the deposited immunoglobulin subclass and the binding pattern seen
However, it provides limited information on the specific antigens that are targeted
Pemphigus diseases (superficial):
Antibodies bind to keratinocytes
Forms a 'chicken wire pattern'
Intercellular deposition of IgG +/- C3

Dermnet- https://dermnetnz.org/topics/pemphigus-vulgaris-pathology/
Pemphigoid diseases (deep):
Antibodies bind linearly to the dermo-epidermal junction (DEJ)
Linear deposition of IgG +/- C3 at the basement membrane zone
Dermnet: https://dermnetnz.org/topics/directimmunofluorescence/
Dermatitis herpetiformis:
Granular or fibrillar deposition of IgA +/- C3 in dermal papillae
Dermnet - https://dermnetnz.org/topics/dermatitis-herpetiformis
Linear IgA disease:
Linear deposition of IgA +/- C3/IgG at basement membrane zone
Intensity of IgA fluorescence higher than IgG and C3
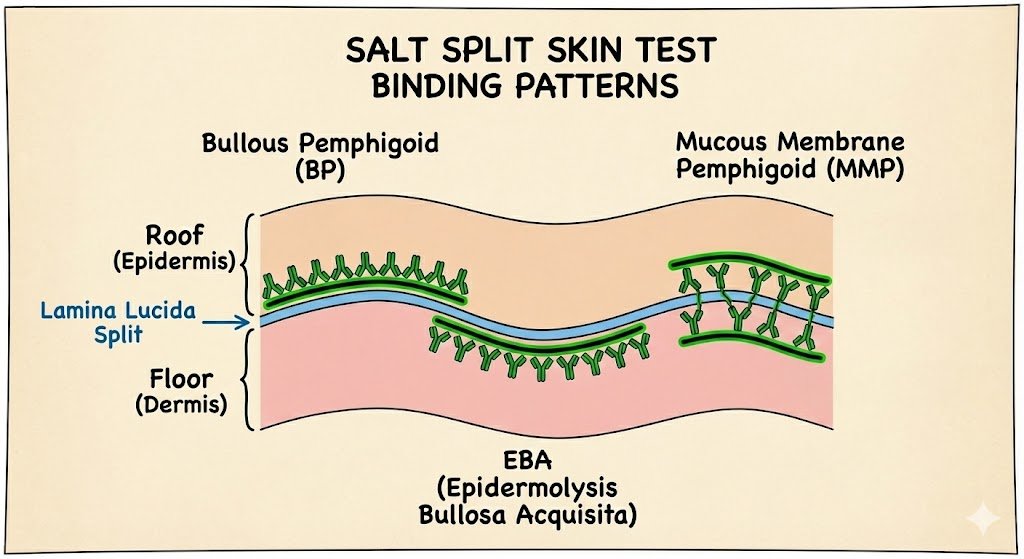
SALT SPLIT SKIN TEST
Helps differentiate bullous pemphigoid from other sub-epidermal blistering conditions.
The biopsy is incubated in 1mol/L salt, which causes a split at the lamina lucida (the 'loose lucida').
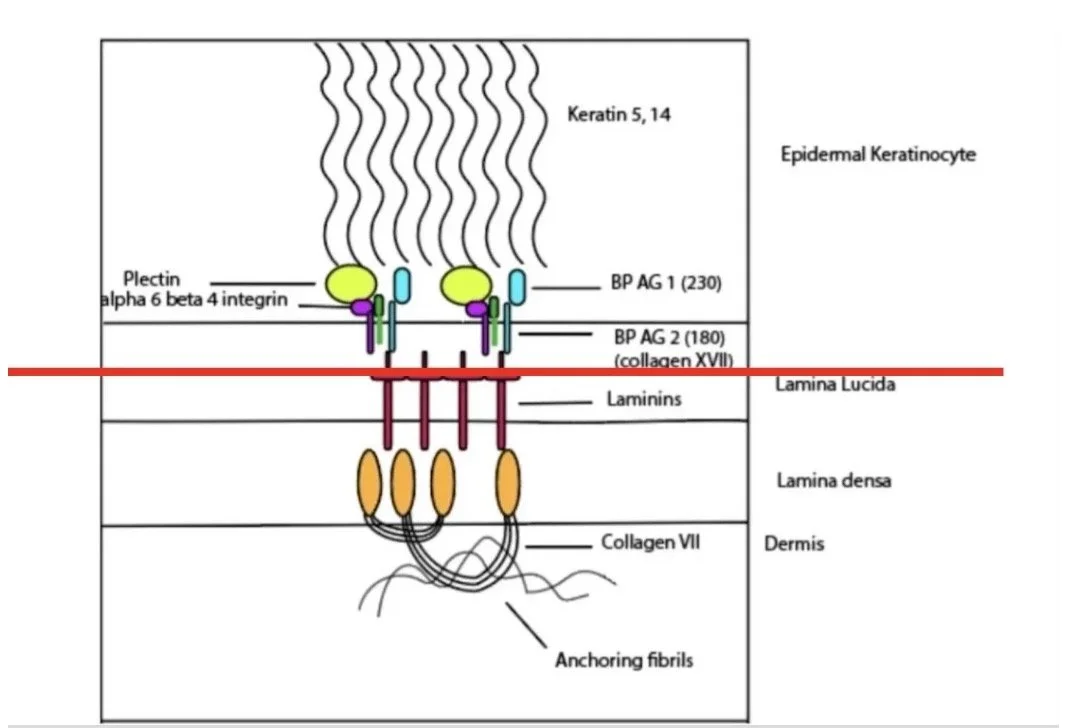
In Bullous Pemphigoid
Antibody (usually IgG) found on the blister roof (epidermal side)
In EBA (Collagen VII):
Antibodies found on blister floor (dermal side)
In Mucous Membrane Pemphigoid:
Deposition can be to roof, floor, or mixed pattern depending on antigens targeted
Roof - BP 180, 230, α6β4 integrin
Floor - Laminin 332, Type VII collagen
SERRATION PATTERN
Looking at the serration pattern refines localization beyond what salt split skin shows.
The salt split separates at the lamina lucida with the following conditions staining on the dermal side:
Laminin γ1 (p200 pemphigoid)
Anti-laminin 332 (associated with mucuous membrane pemphigoid with higher risk of internal malignancy)
Collagen VII (EBA, Bullous Lupus)
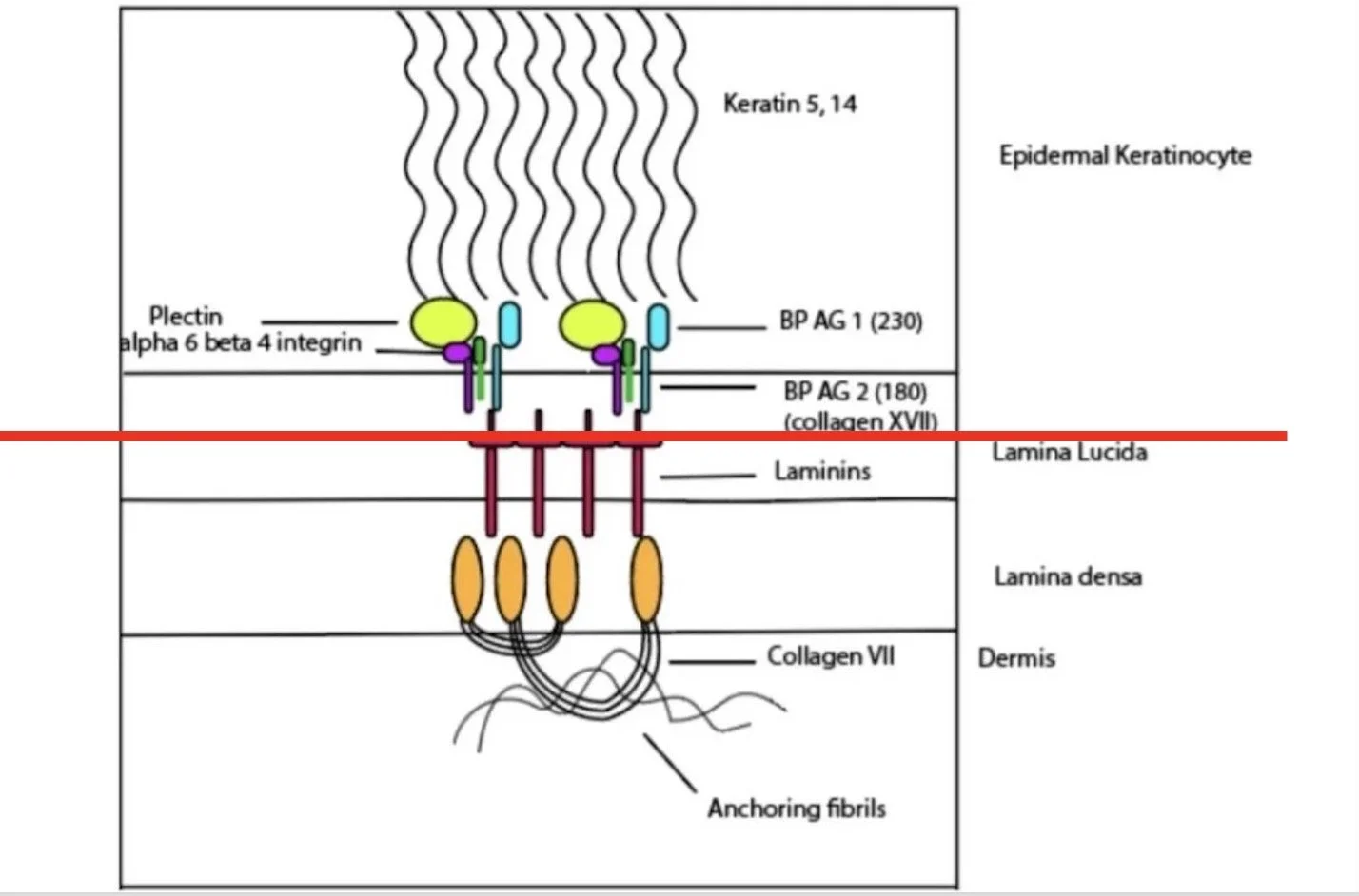
When examining the linear fluorescence on salt split skin at higher magnification, the serration pattern of the undulating basement membrane zone can help distinguish between these deeper conditions
It separates conditions based on whether the antigens at/above the lamina densa or below
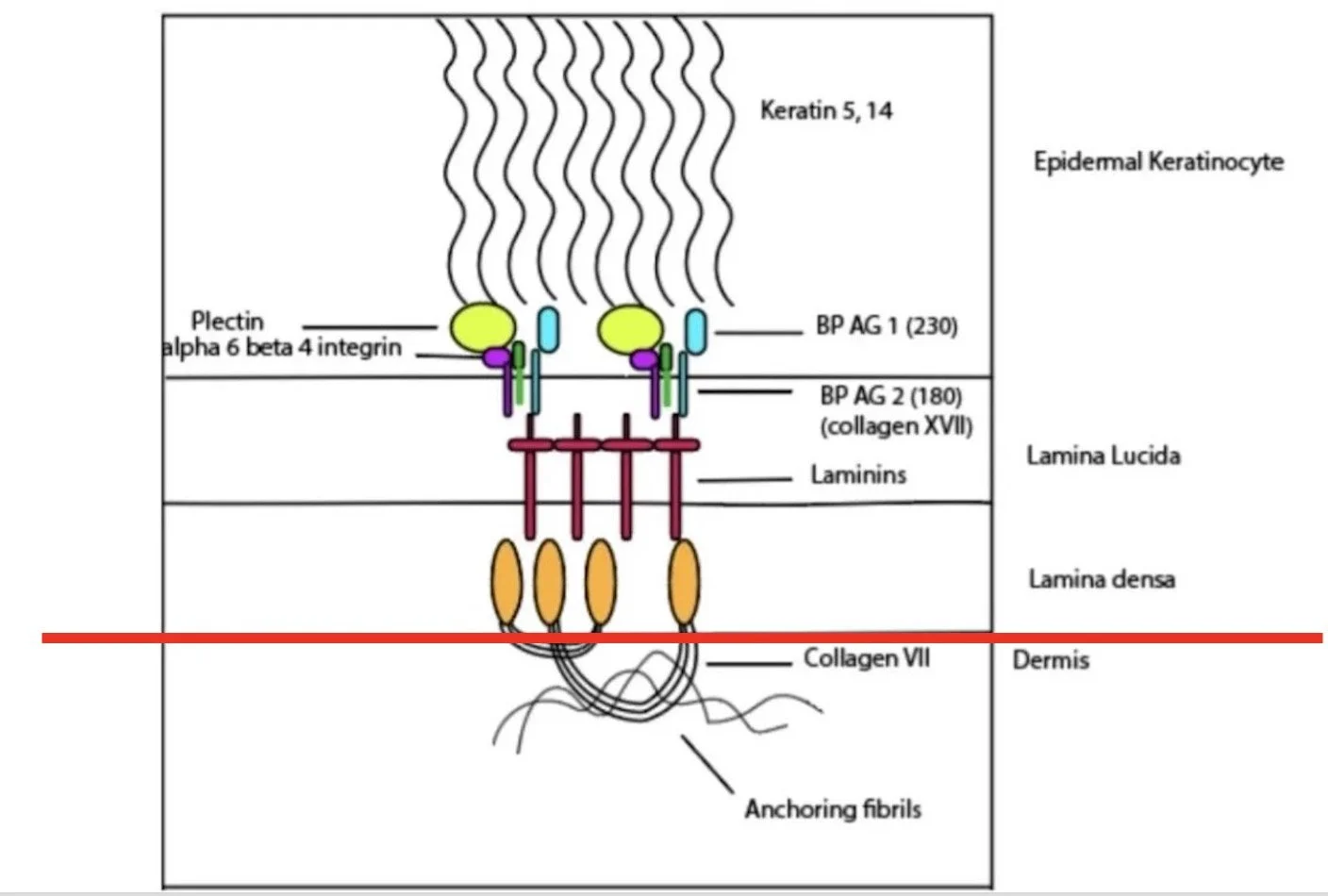
Recap:
Most helpful when salt split shows dermal-side binding (floor) and need to differentiate the conditions further
Memory aid: 'u' for ‘under’ the lamina densa
BLOOD TESTS
Blood tests can be performed for the serologic detection of antibodies
The conventional method performed is:
Initially do indirect immunoflourescence (IIF) microscopy as a screening test
This can be followed by a more antigen-specific serologic assay (eg ELISA) based on the clinical suspicion and IIF screening results
INDIRECT IMMUNOFLOURESCENCE
Detectes circulating antibodies in serum
Very sensitive - good for screening
Not as specific for determining causative antibody as ELISA
IIF is a technique used to detect circulating antibodies in a patient’s serum blood test:
Antibodies/complement (eg IgG, IgM IgA, C3 ) from the patient’s serum (primary antibodies) bind to the target molecules (the substrate) in pre-prepared tissue samples (eg monkey oesophagus)
Then antibodies labelled with dye bind to the above antibodies
The pattern of staining then seen using a fluorescent microscope
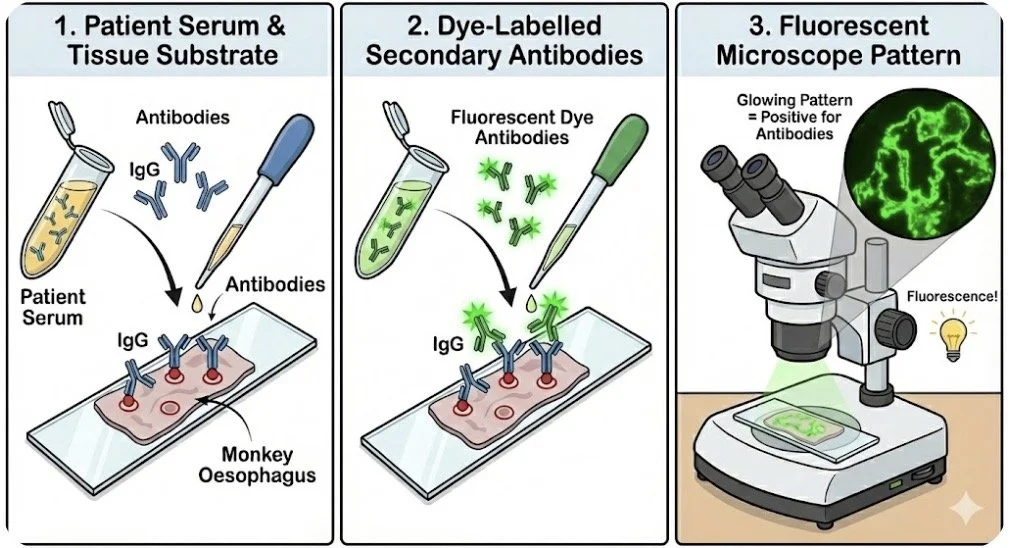
Choice of substrate tissue to use may depend on clinical suspicion of the suspected disease
For instance, classic prepared tissues which are better for certain diseases are:
Sub-epidermal blistering diseases - Normal human skin
Pemphigus vulgaris - monkey oesophagus - monkey oesophagus has high expression of Dsg 3
Pemphigus vulgaris - moneky oesophagus - a monkey acting vulgar
Pemphigus foilaceous - guinea pig oesophagus - good for dsg 1
(think of a guinea pig running in foliage)
Pemphigus foliaceous - guinea pig oesophagus - a guinea pig roaming the foliage
Paraneoplastic pemphigus - rat bladder - plakins (envoplakin, periplakin, desmoplakin) are highly expressed in bladder

ELISA
To achieve a more precise diagnosis, clinicians can identify antibodies directed against specific target antigens using ELISA (Enzyme-Linked Immunosorbent Assay), Immunoblotting, or monospecific Indirect Immunofluorescence (IIF).
ELISA is the most widely used method for targeted antibody detection because it:
Identifies Specific Antigens: It confirms the exact molecular target (e.g., Dsg 1 vs. Dsg 3).
Quantifies Antibody Levels: Unlike standard IIF, ELISA provides a numerical value, making it highly effective for monitoring disease activity and response to treatment.
The following antigens are routinely tested via ELISA at regional centers like St. John’s Institute of Dermatology:
Desmoglein 1 and 3: For Pemphigus variants.
BP180 and BP230: For Bullous Pemphigoid.
Collagen VII: For Epidermolysis Bullosa Acquisita (EBA).
In labs in the USA they can perform more specific tests using specialised techniques
Laminin 332 - Mucosal membrane pemphigoid associated with solid organ cancer
Laminin γ1 - p200 pemphigoid
PEMPHIGUS: AN OVERVIEW
Pemphigus is a group of autoimmune bullous diseases primarily affecting the skin and/or mucous membranes, charachterised by antibody-mediated acantholysis
Acantholysis refers to the separation of keratinocytes due to disruption of intercellular bridges (desmosomes)
Pathophysiology
Keratinocytes are held together by various proteins including proteins called desmosomes. The most clinically relevant desmosomes are desmoglein 1 and 3
Desmoglein 1:
Most abundant in superficial epidermal layers (stratum granulosum/corneum)
Present in high concentrations in skin
Found in lower amounts in mucosal tissues
Desmoglein 3:
Highly abundant in mucous membranes
Present in deeper layers of epidermis (basal and lower spinous) but at lower levels than Dsg1
The Golden Rule: If Dsg 3 is involved, it is Pemphigus Vulgaris.
Pemphigus Foliaceus (Dsg 1 only):
Targets 1 antigen and predmoninantly 1 organ—the skin.
Since Dsg 3 is still working in the mouth, the mucous membranes tend to stay healthy.
Pemphigus Vulgaris (Dsg 3 +/- Dsg 1):
The mucous membranes are usually affected (where Dsg 3 is the primary "glue")
But the skin can also be affected (especially if Dsg 1 affected) and this can also be very severe
Pemphigus Epidemiology
Incidence: 0.5-8 cases per million in Europe
Peak age: 40-60 years (rare under 20 years)
Gender: Slight female preponderance
Ethnicity: Higher frequency in people of Jewish ancestry (particularly Ashkenazi Jews)
Pemphigus vulgaris and foliaceous collectively account for 90-95% of all pemphigus cases
Genetics:
HLA alleles are strongly associated with an increased risk of pemphigus
HLA-DRB104:02 and DQB105:03 most significant
Pemphigus patients may have increased frequency of -
Haematological malignancy: CLL, NHL, Multiple myeloma
Solid organ cancers: oropharyngeal, colon
Autoimmune diseases: Thyroid disease, rheumatoid arthritis
Drugs:
Drugs containing a thiol group have been implicated (penicillamine, captopril)
PEMPHIGUS VULGARIS
Charaterised by antibodies against desmoglein 3 and/or desmoglein 1
The presence of anti-Dsg3 antibodies means mucosal involvement is common and often predominant, as Dsg3 is highly abundant in mucous membranes
When both anti-Dsg3 and anti-Dsg1 antibodies are present, patients tend to experience severe skin disease in addition to mucosal lesions
The acantholysis in PV occurs just above the basal layer of the epidermis, leading to a suprabasilar split
Clinical presentation:
Cutaneous Features:
Shallow, fragile blisters that rupture easily
Leading to erosions and crusting
Initially appear on scalp, face, and shawl-like distribution on upper central torso
Can spread widely
https://dermnetnz.org/topics/pemphigus-vulgaris
Mucosal involvement:
Present at presentation in 50-70% of cases
Increases to 80-90% with time (some series report 100%)
Painful, shallow erosions with irregular borders
Erosive gingivitis common
Most commonly affected intra-oral sites:
Gingiva (80%)
Buccal mucosa (58%)
Palate (26%)
Tongue (15%)
Laryngeal involvement:
Causes hoarse voice, requires urgent ENT review due to risk of oedema and erosions
Ocular involvement:
Crusted erosions on eyelid margins, conjunctival erythema, injection and swelling
Nasal mucosa:
Painful erosions, epistaxis
Genitalia:
Painful erosions
Investigations:
Skin/mucosal biopsy:
For H&E:
Lesional area with bit of blister and surrounding skin
For DIF:
Cutaneous: Peri-lesional area (within 1cm)
Oral disease: can take from normal buccal mucosa (usually sufficiently reliable)
https://dermnetnz.org/topics/pemphigus-vulgaris-pathology
https://dermnetnz.org/topics/pemphigus-vulgaris-pathologyPEMPHIGUS FOLIACEUS
Pemphigus foliaceus results from antibodies primarily directed against desmoglein 1
As Dsg1 is most abundant in superficial layers of the epidermis and found in lower amounts in mucosal tissues, PF typically presents with predominantly cutaneous lesions and rare mucosal involvement
Split occurs high in the epidermis (subcorneal/granular layer)
Skin features:
Crusted and shallow erosions
Often in a seborrhoeic distribution (can sometimes resemble a seborrhoeic dermatitis)
Common areas: scalp, central chest, face, and mid-back
Intact blisters rarely observed (too superficial, rupture quickly)
Erosions generally less painful than PV but can itch/burn


Skin biopsy:
Take a biopsy of a blistered area which includes intact surrounding skin for H&E
Take a biopsy of a peri-lesional area (within 1cm) for DIF
Differential diagnosis intra-epidermal/sub-corneal split on H&E:
Pemphigus foliaceus
IgA pemphigus
Bullous impetigo
Sub-corenal pustular dermatosis
Staphylocccal scalded skin syndrome

Dermnet - indirect immunoflourescence
Endemic Pemphigus (Fogo Selvagem)
Endemic pemphigus, also known as fogo selvagem (meaning 'wildfire' in Portuguese), is essentially an endemic form of pemphigus foliaceus in Brazil
This if thoguht to be environmentally induced possibly from inset bites (eg from black flies).
Proteins left behind from the bite may induce immune mimicry leading to development of antibodies directed against the skin
The investigative findings (H&E, DIF, IIF and ELISA) are consistent with pempihigus foliaceous, showing antibodies against Dsg 1
PARANEOPLASTIC PEMPHIGUS
Paraneoplastic pemphigus is a rare but severe form of pemphigus with a poor prognosis
It is due to an abnormal B or T cell immne respoonse to an underlying malignancy
It is distinguished by antibodies to a wide range of epitopes, including desmoglein 3, desmoglein 1, envoplakin, periplakin, and desmoplakins, among others
Cytotoxic CD8 T cells also play a role which can lead to an interface dermatitis seen alongside acantholysis
This broad range of antibody targets and the cytotoxic T cell role contributes to the polymorphic clinical presentation we often see

Clinical features:
Patients often unwell
Typically begins with painful oral erosions, which can progress to severe and recalcitrant ulceration with pan-stomatitis and hyperplastic mucosa
Haemorrhagic cheilitis (crusted, bleeding lips) and severe ulceration in the oropharynx and genitalia are characteristic and often out of proportion to cutaneous involvement
Haemorrhagic cheilitis and severe oral erosions
The rash is often polymorphic with a variety of lesions and it can mimic other conditions like erythema multiforme, lichen planus or GVHD

Erosions over the torso
https://dermnetnz.org/topics/paraneoplastic-pemphigus
Lichenoid appearing rash on body
https://dermnetnz.org/topics/paraneoplastic-pemphigus
Lichenoid lesions: https://dermnetnz.org/topics/paraneoplastic-pemphigus
Other organs can be affected:
Lung involvement can lead to bronchiolitis obliterans (which can be severe)
GI tract can be involved
Associated cancers:
Lymphoma ≈ 40% of cases
CLL ≈ 30%
Castleman’s disease ≈ 10%
Castleman disease is a rare disorder that involves an overgrowth of cells in body’s lymph nodes
Most common form of the disorder affects a single lymph node (unicentric Castleman disease) usually in chest or abdomen
Multicentric castleman disease affects multiple lymph nodes throughout the body and has been associated with HHV 8 and HIV
Thymoma ≈ 6%
Spindle cell neoplasms ≈ 6%
Waldenstrom’s macroglobulinaemia ≈ 6%
Investigations

Dermnet - pareneoplastic pemphigusDermnet - paraneoplastic pemphigusManagement
To do….
PEMPHIGUS VEGETANS
Pemphigus vegetans is a rare variant of pemphigus vulgaris (approx 2% of cases) which is chracterised by its clinical presentation rather than distinct antibody profiles
Clinically patients get vegetating, hyperkeratotic, papillomatous and fissured erosions
It typically affects flexural areas such as the axilla, groin and under the abdomen
They are often bacterial colonised and malodorous
Oral lesions are common and patients can get a distinctive cerebriform tongue
H&E: Histology may show eosinophilic abscesses or spongiosis with few acantholytic cells, which might not immediately look like typical pemphigus
DIF: Shows characteristic intercellular depoisition of IgG (chickenwire)
IIF: ELISA shows antibodies against Dsg 3 and desmocollins
PEMPHIGUS ERYTHEMATOSUS
(Also known as Senear-Usher syndrome)
Is a localised form of pemphigus foilaceous
Get scaly and crusted lesions across the malar area of the face and in other seborrhoeic areas
May remain localised for years or evolve into a more generalised pemphigus foilaceous
It is called pemphigus erythematosus as it shares immunologic features of both lupus erythematosus and pemphigus
Get IgG and C3 seen on cell surfaces of keratinocytes and in the basement membrane zone
May have serologic findings suggestive of SLE especially positive ANA
IgA PEMPHIGUS
There are two types of IgA pemphigus:
1. Subcorneal pustular dermatosis type (SPD)
2. Intraepidermal neutrophilic type (IEN)
Desmocollin 1 is the main antigen target overall particularly in the SPD sub-type
Dsg 1 and 3 can be targeted also
Both types associated with intercellular IgA deposits on immunoflourescence
There is NO ACANTHOLYSIS on histology (unlike pemphigus vulgaris, pemphigus foilaceous and paraneoplastic pemphigus)
Clinical:
SPD clinically presets as serpiginous vesicles and pustules
IEN classically presents as flaccid pustules and bullae arranged in an annular, polycyclic (sometimes referred as sun-flower like) arrangements usually in intertriginous areas
The pustules tend to quickly rupture and you get an annular crust over the plaque
Commonly affects flexural areas like axilla and groin but can be more widespread
Dermnet - IgA pemphigusAssociations:
Monoclonal IgA gammopathy
Multiple myeloma
Others reported: HIV, Sjogrens, RA, Crohn’s
Investigations:

H&E:
Intra-epidermal neutrophillic infiltration
Little to no acantholysis - have mild loss of cohesion between keratinocytes
Subcorneal pustular dermatosis subtype - increased intensity of IgA autoantibodies in the upper surface of the epidermis.
Intraepidermal neutrophilic - inflammatory infiltrates, primarily in either the entire or lower part of the dermis.
DIF:
IgA on epidermal cell surfaces
IIF:
Positive circulating IgA
Treatment options:
Steroids: topical and oral (0.5-1mg/kg)
Addition of Dapsone far superior to just steroids alone (dapsone often good for treatments with neutrophilic infiltrates)
Other agents: Colchichine, sulfapyridine, acitretin, PUVA
Less often: MMF, adalimumab (TNf-alpha is involved in activation of neutrophilic infiltration of epidermis so may hinder further progression of IgA pemphigus)
Ddx:
Subcorneal pustular dermatosis (aka Sneddon Wilkinson disease)
Sterile pustular lesions that erupt in cyclical patterns
Like IgA pemphigus the pustular lesoins coalesce in an annular pattern and burst to form crusted plaques
Similar area of distribution - groin, trunk, axilla and avoid mucous membranes
Histology shows perivascular infiltration of neutrophils and spongiosis
DIF negative for IgA
Pemphigus foliaceous
DIF is key - pemphigus foliaceous demonstrates IgG to DsG 1
Bacterial skin infections
Linear IgA Disease
Prognosis:
Much milder and limited disease compared to classic pemphigus
MANAGEMENT OF PEMPHIGUS
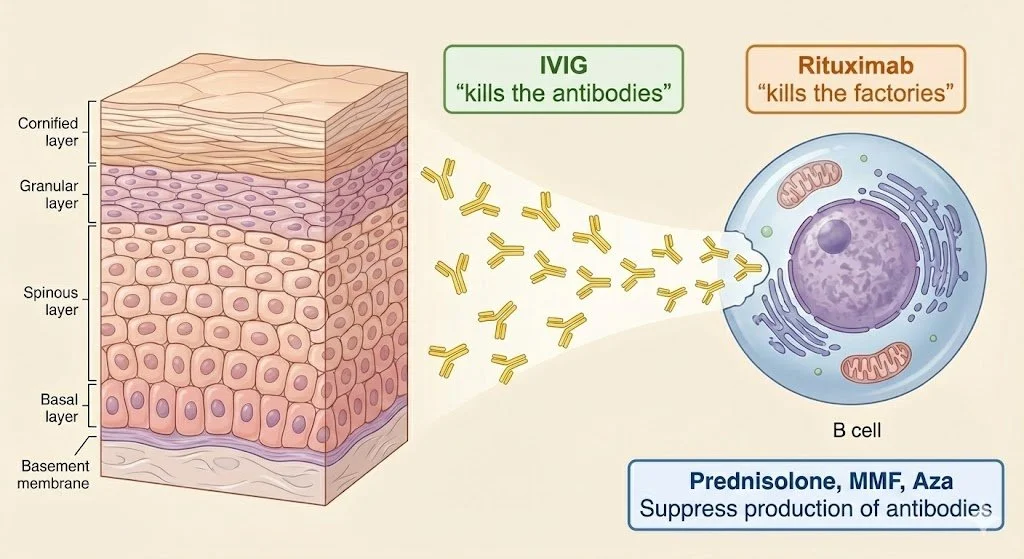
In pemphigus, B cells produce antibodies that target desmogleins, which are proteins that connect epidermal cells together. When considering treatment options, it's helpful to think of B cells as factories producing pathogenic antibodies:
Rituximab is an anti-CD20 antibody that directly targets B cells
Essentially 'destroys the factories' producing harmful antibodies
Other immunosuppressive agents (prednisolone, MMF, azathioprine) reduce the rate of antibody production by these cellular factories
IVIG promotes the destruction of circulating antibodies that have already been produced
There are UK and European guidelines regarding the management of pemphigus:
Enk A, et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2020 Sep;34(9):1900-1913. doi: 10.1111/jdv.16752.
Harman KE, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. 2017 Nov;177(5):1170-1201. doi: 10.1111/bjd.15930.
The key difference is the 2020 European guidelines recommend rituximab combined with tapering corticosteroids as a first-line treatment for moderate and severe pemphigus, whereas the 2017 British guidelines classify rituximab as a third-line treatment for refractory disease (however this was published prior to key trials demonstrating Rituximab’s efficacy as a first line treatment)
I attended a few clinics in St. John’s Institute Immunbullous clinic in 2024 and in general their practice was to trial a conventional systemic agent (eg MMF) for 3-6 months prior to commencing Rituximab
Please see https://www.stjohnsdermacademy.com/podcasts for a good reference podcast on Pemphigus vulgaris
I will discuss what they tend to do in St. John’s going forward but may be argument in Ireland to use Rituximab first line for moderate to severe pemphigus as per European guidelines
Initial skin care:
Topical agents
Antiseptic soap substitute if erosions present - Octenisan wash once daily (avoid Dermol 500 which is often used in UK due to increased irritant reactions)
Face - Moderate potency steroids (eg eumovate)
Body - Potent/super potent steroids (eg dermovate)
Oral agents
Betamethasone Solution:
Betamethasone 500mcg tablet dissolved in 10-20ml water
Use as mouthwash for 5 minutes up to QDS
Do not swallow
Flixonase Nasules:
Fluticasone propionate 400mcg contents in 10-20ml water
Mouthwash for 5 minutes up to QDS
Dermovate in Orobase Gel:
Apply directly to affected areas
Better for deeper, persistent lesions
Candida Prevention:
Add 1ml nystatin to steroid mouthwashes OR use standard antifungal therapy
Low threshold for treating oral candida
Systemic therapies for pemphigus:
Systemic Corticosteroids:
BAD Guidelines: Up to 1.5mg/kg daily
St. John's Approach: 0.5mg/kg daily (to minimise long-term exposure with associated complications)
Example Regimen (60kg patient):
Start: Prednisolone 30mg daily
Reduce by 5mg every 2 weeks down to 20mg
Slower reduction: 2.5mg every week/fortnight down to 10mg
Very slow reduction: 1mg weekly down to 5mg maintenance
Once reducing below 5mg need to consider a 9am cortisol +/- short synacthen test with gradual wean over severeal months to allow adrenal recovery
Ensure PPI and bone protection
Can consider pulse methylprednisolone in severe disease: 250-500mg IV daily for 2-5 days or as an adjunct to IVIG or rituximab
Conventional systemic agents:
Conventional immunosuppressive agents that tend to be used in pemphigus are azathioprine and mycophenolate mofetil
Mycophenolate mofetil often used due to increased risk skin cancer associated with long-term exposure to azathioprine (azathioprine possibly has marginal superiority based on head to head trials)
Mycophenolate mofetil:
Dosing:
Start: 500mg BD
Increase to 1g BD within 2-4 weeks
Maximum: 2g daily (3g daily shows no superiority with more side effects)
Monitoring:
Pre-treatment: FBC, U&E, LFTs, pregnancy test, hepatitis B/C, HIV
Ongoing: Weekly × 2 weeks after dose increases, then 8-12 weekly
Side Effects: Diarrhoea, nausea, infection, myelosuppression, hepatotoxicity, severe teratogenicity
Clinical Evidence:
Approximately 70-80% sustained response
Delayed onset (≥4 months)
Switch to Myfortic if GI side effects
1g MMF = 720mg Myfortic
Azathioprine:
Dosing: 1-2.5mg/kg daily
Essential: TPMT level pre-treatment
Safe during pregnancy
Other agents (Limited evidence):
Dapsone - One RCT showed slight steroid-sparing effect
Tetracyclines - Limited evidence, adjunctive use only
Methotrexate - Retrospective studies, weak evidence
Rituximab:
Rituximab (anti-CD20 monoclonal antibody) works by destroying B cells, effectively eliminating the "factories" responsible for producing the harmful autoantibodies that cause pemphigus.
Indications:
Licensed for pemphigus vulgaris
European Guidelines: Favour earlier use as first-line therapy
Consideration in UK:
Failure of MMF/Azathioprine after 3-6 months
Unacceptable prednisolone dose (>7.5mg daily)
Clinical evidence - RITUX-3 Trial (First-line use):
90 patients with PV/PF
Rituximab + short-term prednisolone (3-6 months) vs standard prednisolone (12-18 months)
Primary endpoint met: Complete remission without steroids at 24 months
Superior efficacy with fewer adverse events
Complete remission: 89% vs 34%
Relapse rates: 42% vs 83%
Standard Protocol:
1g × 2 infusions, 2 weeks apart
Licensed for additional 500mg at 12 and 18 months, then 6-monthly
St. John's Practice: Base repeat dosing on clinical/immunological picture
Pre-medication:
IV methylprednisolone 100mg
Paracetamol
Chlorphenamine
Monitoring:
B-cell counts and antibody titres
B-cell repopulation predicts clinical relapse
Contraindications:
Severe active infection
Severe heart failure
Side Effects:
Infusion reactions (headache, fever, nausea, chills)
Increased infection risk (HSV, VZV, respiratory, UTI)
Hepatitis B reactivation
Progressive multifocal leukoencephalopathy (extremely rare in AIBD)
Covid-19 considerations and vaccination Strategy:
Critical: Vaccinate 2 weeks prior to first rituximab dose
Significantly impaired vaccination response for ≥1 year post-treatment
Increased risk of severe COVID-19 infection
Consent patients regarding COVID morbidity/mortality risk
Adjuvant systemic agents considerations:
St. John’s half the MMF dose (eg 1g bd to 500mg bd) when commencing rituximab to avoid excessive immunosuppression
The dose is reduced on the first day of the infusion
ADVANCED AND RESCUE THERAPIES
IVIG (intravenous immunoglobulin)
Mechanism:
Intravenous immunoglobulin (IVIG) works by providing a high concentration of donor antibodies that neutralize and accelerate the clearance of the patient's own harmful autoantibodies, thereby preventing them from damaging the skin's cell-to-cell bonds.
Dosing: 2g/kg divided over 3-5 days monthly × 3 cycles
Pre-screening:
Baseline immunoglobulins (exclude IgA deficiency)
If given IVIG with IgA deficiency can develop antibodies to IgA and develop anaphylaxis
Baseline hepatitis serology
When receive IVIG can get passive transfer of antibodies - such as HBcAb, HBsAb, anti-HCV
But antigens should will remain negative - eg anti HBsAg will remain negative and HBV DNA (PCR) will remain negative
Assess thrombosis risk
Uses:
Severe acute disease (inpatient setting)
"Bridging" therapy while awaiting immunosuppression effect
Patients where immunosuppression risky (malignancy/infection)
Cyclophosphamide:
Evidence: 85% disease control in severe PV (oral 2-2.5mg/kg)
Dosing Options:
Oral: 2-2.5mg/kg daily
Pulsed IV: 500mg monthly + 3 × IV methylprednisolone 500mg
Low-dose oral: 50mg OD between pulses
Limitations: Myelosuppression, alopecia, haemorrhagic cystitis, fertility issues
Plasma Exchange:
Removes pathogenic immunoglobulins + beneficial proteins
Risk of rebound antibody production
RCT in newly diagnosed patients: no benefit demonstrated
Immunoadsorption:
Selective removal of immunoglobulins
Preserves useful plasma components
Expensive, limited access
Reserved for refractory cases
MONITORING AND FOLLOW-UP
Clinical assessment:
Control: No new lesions forming, existing lesions healing
Remission: Complete healing maintained for ≥6 months
Serological monitoring:
Desmoglein 1 and 3 ELISAs at each visit
Titres correlate with disease activity
Normalised antibodies: Much lower flare risk
Guide for treatment withdrawal
Steroid Withdrawal Protocol
Clinical remission for 6-12 months
Serological remission (normal/significantly reduced antibodies)
Gradual MMF wean first
9am cortisol before reducing prednisolone <5mg
Slow steroid taper with endocrine input if needed
Bone Protection
Baseline DEXA essential
FRAX score: Sheffield.ac.uk/FRAX
NOGG guidelines: nogg.org.uk/full-guideline
Bisphosphonates if above intervention threshold
All patients: adequate calcium and vitamin
Management pemphigus sub-types
Management of Pemphigus Foliaceus
Management follows the same treatment ladder as pemphigus vulgaris, tailored to disease severity.
As pemphigus foliaceus lacks mucosal involvement, patients are often less severely affected overall.
Topical corticosteroids may be sufficient for limited disease. For more widespread disease, oral prednisolone with a steroid-sparing agent (typically MMF) is used, with escalation to rituximab for refractory cases as needed.
Management of Paraneoplastic Pemphigus
Treatment of the underlying malignancy is essential
The prognosis is generally poor, with reported one-year mortality rates of up to 90%.
This is attributable to the aggressive mucocutaneous disease, the associated underlying neoplasm (commonly NHL, CLL, Castleman's disease, or thymoma), and the potential for developing bronchiolitis obliterans.
Immunosuppressive therapy follows similar principles to pemphigus vulgaris, but responses are often limited if the underlying malignancy cannot be controlled.
DEEP BLISTERING
If the split occurs in the basement membranze zone the blisters will be more tense
The basement membrane zone is complex with multiple proteins within it
Different clinical conditions occurs due to lack of specific proteins through attack or congenital lack of these proteins (epidermolysis bullosa congenita)
BULLOUS PEMPHIGOID
Introduction
The most common immune blistering disease
Due to antibodies (predominantly IgG) binding to targets in within the DEJ
BP 180 (aka BP Ag 2/Collagen XVII) - commoner
BP. 230 (aka BP Ag1)
This leads to the formation of sub-epidermal bullae
Epidemiology
Predominantly a disease of the elderly
Can affect any age and paediatric cases have been reported
Genetics:
There may be a genetic tendency towards the disease and there have been a few HLA associations identified (DR-B1, DQ-A1, DQ-B1)
Drugs:
Dipeptidyl-4 drugs (aka DPP-4 or gliptins) are associated with a x 3 fold increase in developing the disease
Other potential triggers - loop diuretics, thiazide diuretics, aldosterone antagonists
Neurological disease:
Patients with bullous pemphigoid (BP) have a recognised increased prevalence of neurological disease.
Dementia
Multiple sclerosis
Parkinson’s disease
Stroke
Epilepsy
This association is supported by the presence of neural isoforms of BP230 in both the central and peripheral nervous systems.
Clincial presentation
Pre-bullous phase of BP:
Often get a prodromal phase which can occur for weeks or months before the onset of blistering
Usually characterised by urticated erythema (lesion appear like hives)
Patients can just have a generalised pruritius so in pruritic elderly patients without a rash you can consider indirect immunoflourescence for BP 180/230 if no other cause is found
Bullous phase of BP:
Patients then develop tense fluid-filled bullae on normal skin or erythematous base
Fluid tends to be serous or can be haemorrhagic
Bullae can be widespread, mainly on trunk and limbs
Blisters can be intensely itchy
Bullae rupture to leave erosions which heal without scarring (although there may be persistent erythema)
Milia formation is rare which differentiates it from other sub-epidermal diseases


Oral BP:
Affects mucosal surfaces in only 10% of patients
Tend to be shorter in duration than MMP and non-scarring so if they are persistent and difficult to treat consider MMP
Investigations
Dermnet - https://dermnetnz.org/topics/bullous-pemphigoid-pathologyDdx: Sub-epidermal split with eosinophils:
Bullous pemphigoid
Bullous drug eruption
Bullous arthropod bite
Prognostic factors in bullous pemphigoid:
Treatment can be tricky as patients usually elderly (mean age 80)
Often have poor general health
Deletrious prognostic factors:
Older age
Cardiac insufficiency
Dementia
Past history of stroke
Poor general condition
Use of high doses of corticosteroids
Management
There are currently two major sets of guidelines regarding the management of bullous pemphigoid (BP):
• Venning VA, et al. British Association of Dermatologists' (BAD) guidelines for the management of bullous pemphigoid 2012. Br J Dermatol.
• Borradori L, et al. Updated S2K guidelines for the management of bullous pemphigoid initiated by the European Academy of Dermatology and Venereology (EADV). 2022. J Eur Acad Dermatol Venereol.
One key difference between these documents lies in the approach to systemic corticosteroid dosing
The 2012 BAD guidelines historically recommended a stratified approach to oral prednisolone based on disease severity, suggesting 0.75–1.0 mg/kg for severe cases, 0.5 mg/kg for moderate disease, and 0.3 mg/kg for mild or localised cases3. However, the 2022 European (EADV) guidelines now recommend a standard starting dose of 0.5 mg/kg/day across all severity levels (mild, moderate, and severe)45.
This shift is based on evidence that high-dose regimens (1.0 mg/kg) are associated with significantly higher mortality and treatment-related complications in the elderly population typically affected by BP. Current practice at St. John’s Institute of Dermatology—whose experts contributed to the 2022 European update—aligns with this safer, moderate-dose starting point
As with pemphigus management I will discuss the general approach that was taken in St. John’s although practices may differ elsewhere
General measures
General skin care principles:
Antibacterial soap substitutes: Octenisan wash once daily (lather all over, leave for 5 minutes, wash off, pat dry)
Emollient: Emulsifying ointment or something similar
Non-adherent dressings: Mepitel/Mepilex as appropriate
Potassium permanganate soaks: Helpful for wet, weepy erosions that need drying out
Blister management:
Gently cleanse blister with antimicrobial solution, taking care not to rupture
Pierce blister at base with sterile needle/syringe (bevel facing up)
Select a site where fluid will drain by gravity to discourage refilling
Gently apply pressure with sterile gauze to facilitate drainage
DO NOT DE-ROOF THE BLISTER!
After drainage, cleanse again with antimicrobial solution
First line agents
Can consider topical steroids and doxycycline 200mg daily in certain cases if on the milder spectrum and patient has many co-morbidities and is at higher risk from systemic steroid complications
Many patient however will require oral steroids to induce remission and some will require second line immunosuppressive agent or dapsone
Topical steroids:
Dermovate (clobetasol propionate) ointment to eroded and erythematous areas on body
Eumovate ointment to affected areas on the face
Tetracycline antibiotics:
Doxycyline 200mg daily (eg 100mg bd)
Used in bullous pemphigoid for their anti-inflammatory effects (not anti-microbial)
Many clinicians commence doxycycline alongside topical or oral prednisolone as an adjunct
Stomach protection is important especially if considering systemic steroids as both drugs can cause gastritis/oesophagitis
Evidence for use of doxycycline in bullous pemphigoid is noted below
BLISTER Trial Evidence (2017 UK RCT)
Compared doxycycline 200mg daily vs prednisolone 0.5 mg/kg in 121 patients
At 6 weeks: Prednisolone more effective for short-term blister control (91% vs 74% achieving ≤3 blisters)
BUT doxycycline met non-inferiority margin and had better safety profile
Conclusion (from study): Doxycycline is a reasonable first-line option, especially when minimising corticosteroid exposure is important
Oral steroids:
The aim is to try to start at the lowest possible dose that might control the disease
Caution to be applied in patients with diabetes, hypertension or poor bone health
In patients with moderate to severe disease reasonable to start at 0.5mg/kg
Maintain this till have control of disease activity - defined as where no new lesions are forming and existing lesions are beginning to heal
Then begin to wean
Second line agents
Consider adding a second line agent if the disease starts to flare on the reduction of prednisolone
Mycophenolate mofetil:
In general prepared to azathioprine due to risk of skin cancer associated with long term azathioprine use
Dosing: Start 500mg bd and increase to 1g bd within a couple of weeks
Max dose: 2g daily (3g not superior and associated with more side effects)
Side effedcts: GI disturbance (dose-dependenant, up to 20% ag 2g daily)
Myfortic (mycophenolic acid) is an alternative if have GI side effects with MMF
Contraindicated in pregnancy (severe teratogenicity)
Azathioprine:
Roughly equivalent clinical efficacy to MMF
TPMT check essential before starting
Key concern: Increased risk of skin cancer (SCC) with long-term use
Considered safe during pregnancy
Methotrexate:
Generally evidence base less than MMF/Azathioprine
If used, consider using a lower dose (5-12.5mg weekly subcut) due to the increased risk of complications in an elderly population
Dapsone:
Can be considered in patients who have contraindications to oral steroids and other immunosuppressive treatments in mild to moderate bullous pemphigoid
Be careful prescribing dapsone in older patients with cardiovascular disease (as they would be more sensitive to any anaemia that could occur with dapsone)
Check G6PD before starting
Side effects: Haemolytic anaemia, nausea, headache, methaemoglobinaemia, peripheral neuropathy (predominantly motor)
Rituximab:
Data suggests it is not as rapidly or completely effective for pemphigoid as it is for pemphigus
But can be considered in refractory pemphigoid
IVIG:
Can be very helpful in an acute setting – eg hospitalised patients
• 2g/kg monthly typically split over 3-5 days
• +/- methyl prednisolone 250–500mg daily ×3 (to help speed up response)
• Repeat for at least 3 cycles
Cyclophosphamide:
Generally would be more likely to try rituximab first prior to cyclophosphamide
Dupilumab:
LIBERTY-BP ADEPT Trial (Phase 2/3 RCT, n=106)
Press release - not fully published yet
First positive RCT for a biologic in BP
Sustained remission at week 36: 20% vs 4% placebo (both arms received tapering steroids)
Significant steroid-sparing effects noted
Safety comparable to placebo; conjunctivitis more common with dupilumab
https://pubmed.ncbi.nlm.nih.gov/38443648/
Murrell DF et al. Study Design of a Phase 2/3 Randomized Controlled Trial of Dupilumab in Adults with Bullous Pemphigoid: LIBERTY-BP ADEPT. Adv Ther. 2024;41(7):2991-3002.Retrospective Cohort Study (n=103, 34 hospitals in Spain)
53% complete response by week 4; 96% clear by week 52
80% reduction in corticosteroid use
https://pubmed.ncbi.nlm.nih.gov/39418120/
Melgosa Ramos FJ, et al. Real-world evaluation of the effectiveness and safety of dupilumab in bullous pemphigoid: an ambispective multicentre case series. Br J Dermatol. 2025.Impression -
Dupilumab has RCT-level evidence demonstrating efficacy in BP with rapid real-world responses.
Omalizumab:
French Observational Multicentre Study (n=100)
All patients refractory to ≥1 treatment line
Complete remission ahieved in 77%
Median time to complete remission: 3 months.
https://pubmed.ncbi.nlm.nih.gov/37792727/
Chebani R, Lombart F, Chaby G, et al. Omalizumab in the treatment of bullous pemphigoid resistant to first-line therapy: a French national multicentre retrospective study of 100 patients. Br J Dermatol. 2024;190(2):258-265.Systematic Review (n=56)
Overall response: 87.5% (55% CR, 32% PR)
57% relapsed on discontinuation but most recaptured response
https://pubmed.ncbi.nlm.nih.gov/35379011/
D'Aguanno et al. Omalizumab for the Treatment of Bullous Pemphigoid: A Systematic Review of Efficacy and Safety. J Cutan Med Surg. 2022;MUCOUS MEMBRANE PEMPHIGOID
Mucous membrane pemphigoid (MMP) is a mucocutaneous blistering disease characterised by antibodies against components of the dermo-epidermal junction (DEJ) with predominant mucosal involvement.
It is much rarer than bullous pemphigoid
Incidence: approximately 2 per million per year
Mean age of onset: 60-65 years
Female preponderance: 1.5-2.3:1
Can affect multiple mucosal sites with potential for scarring
MMP is mediated by autoantibodies targeting multiple basement membrane zone antigens.
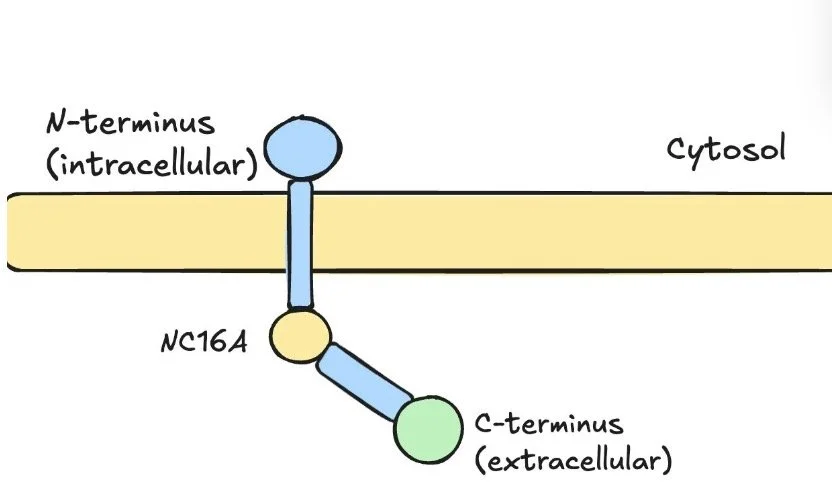
Unlike bullous pemphigoid where the NC16A domain of BP180 is predominantly targeted, MMP often involves antibodies to the C-terminal domain of BP180
Standard ELISAs miss the C-terminal domain so might not come up positive in MMP
Clinical presentation
Oral involvement (85%)
Usually the presenting feature and frequently are the most challenging to manage
Shallow blisters/erosion palate
Desqaumative gingivitis
Dermnet - https://dermnetnz.org/topics/mucous-membrane-pemphigoid
Most common intraoral sites:
Gingiva (80%)- commonest and often presents as a desquamative gingivitis
Buccal mucosa (approx 60%)
Palate (25%)
Alveolar ridge (15%)
Tongue (15%)
Tend to get a chronic erosive gingivitis wiht painful ulceration
Intact blisters are rarely seen
Lesions heal with scarring (unlike in oral BP)
Ocular involvement (65%):
Ocular involvement requires aggressive treatment due to potential for blindness
Adhesions between bulbar mucosa and conjunctiva
Clincial progression:
Early: Conjunctival erythema, irritation, foreign body sensation, mucous discharge
Intermediate: Shortening of inferior fornix, symblepharon formation (adhesions between conjunctival surfaces)
Late: Trichiasis (eyelashes pointing in), entropion (inward angulation of eyelid), lagophthalmos (abnormal eye closure)
End-stage: Corneal scarring leading to blindness
Clinical pearl: β4 integrin antibodies are particularly associated with ocular lesions.
Anogenital invovlement (20%)
Erosions and scarring can mimic lichen sclerosus
Results in architectural abnormalities
Vaginal stenosis may occur in women
Laryngeal/Pharyngeal/Oesophageal Involvement (10-20%)
Voice hoarseness (laryngeal involvement)
Odynophagia
Patients with hoarse voice need urgent ENT review
Cutaneous Involvement (25-30%)
Lesions resemble bullous pemphigoid
Tense bullae on normal or erythematous skin
Heal with scarring and milia formation (unlike BP)
Scalp involvement can cause scarring alopecia
Laminin 332 MMP:
Associated with severe MMP phenotype
25-30% association with solid organ malignancy
All patients with base-binding pattern on salt-split skin should have CT chest/abdomen/pelvis
Can’t test for laminin-332 with ELISA in Ireland/UK
Investigations
H&E:
Subepidermal blister with mixed inflammatory infiltrate (similar to BP)
But often can be a ‘cell-poor’ blister
Tends to have fewer eosinophils than bullous pemphigoid
See fibrosis and scarring in later stages
Histology alone cannot distinguish MMP from BP, EBA or Linear IgA disease
DIF:
Linear deposition of IgG, IgA and/orC3 at BMZ
IgA deposition more prevalent in MMP than BP
IIF:
Interestingly, IIF can be is lower yield for MMP compared to Bullous Pemphigoid so you are more likely to have a negative test
Salt split skin testing:
In MMP antibodies can bind to the roof or the floor
If binds to the floor you might think that laminin-332 is being targeted so consider ordering a CT chest/abdo/pelvis (? underlying malignancy)
ELISA:
Some labs may be able to test for anti-BP 180 carboxy terminal domain and anti-laminin-332
Management
MMP is generally more challenging to treat than bullous pemphigoid. It typically follows a chronic relapsing course, and complete remission is less common than in BP. The presence of dual IgG and IgA antibodies at the DEJ is a poor prognostic indicator.
First Line
Topical corticosteroids (superpotent to affected areas)
Corticosteroid mouthwashes (betamethasone 500mcg dissolved in water, or fluticasone nasules)
Tetracycline antibiotics (doxycycline 200mg daily)
Dapsone
Second Line
Oral prednisolone (0.3–0.5mg/kg) combined with:
Mycophenolate mofetil (MMF) or azathioprine
Continue dapsone as adjunctive therapy
Third Line
Rituximab (1g × 2, two weeks apart)
Cyclophosphamide
IVIG (2g/kg over 3–5 days)
Ocular involvement requires aggressive treatment — collaborate closely with ophthalmology
MMP often requires combination therapy (e.g., prednisolone + dapsone + immunosuppression) to achieve disease control
Dapsone plays a role in the management of MMP due to the role of neutrophils in the subepidermal blistering
Cicatricial pemphigoid:
Used to be used interchangeably with MMP
Ideally should only be used for rare events where you have pemphigoid that causes scarring but does not affect mucous membranes (but in practice cicatricial pemphigoid and MMP are probably still used interchangably)
Need to exclude other sub-epidermal blistering conditions (EBA, MMP) prior to making diagnosis of cicatricial pemphigoid
Brunsting-Perry cicatricial pemphigoid:
Is a sub-type of pemphigoid presenting as blisters, erosions and crust on the head and neck
These tend to heal with scarring
Antibodies targeted include BP-180, BP-230 and to a lesser extent Collagen VII and laminin-332
PEMPHIGOID GESTATIONIS
Blistering disorder that can occur during pregnancy
Postulated that HLA mismatch between mother and foetus may trigger an immune response that cross-reacts with maternal skin
Characterised by IgG antibodies directed against BP 180 in the BMZ
Antibodies to BP230 can be present in about 10%, the significance is unclear
Clinical features:
Usually presents in a prebullous phase involving urticarial plaques and papules on the abdomen that characteristially involve the periumbilical skin (may start here)
The rash may then spread to the rest of the torso, the intertriginous areas and extremities
The face, palms and soles usually spared
Mucosal involvement occurs in less than 20% of cases
Dermnet - https://dermnetnz.org/topics/pemphigoid-gestationisDermnet - https://dermnetnz.org/topics/pemphigoid-gestationisCan occur any time between 4 weeks gestation and 5 weeks postpartum with the majority presenting in the 2nd and 3rd trimester
Onset in 1st or 2nd trimester can lead to adverse pregnancy outcomes:
Decreased gestational age at delivery
Preterm birth
Low birth weight
But overall it has a generally good foteal prognosis
Can be associated with other autoimmune diseases in the mother (eg Grave’s disease) and more rarely with trophoblastic tumours (choriocarcinoma, hydatidiform moles)
H&E:
Identical to bullous pemphigoid
DIF:
Linear C3 in all cases, IgG only seen in approx 25% of cases
ELISA:
Antibodies to NC16a domain of BP 180 seen(very sensitive and specific)
Management
Case reports and small case series have suggested Dupilumab could also be effective in the management of pemphigoid gestationis
LINEAR IgA DISEASE
Linear IgA bullouse dermatosis (LABD) is a sub-epidermal blistering disease characterised by linear deposition of IgA at BMZ
It is the commonest autoimmune blistering condition in children
Epidemiology:
Bimodal age distribution:
Childhood: peak at approximately 5 years (chronic bullous disease of childhood)
Adult: peak at 60-65 years
Slight female preponderance
Can be idiopathic or drug-induced
Drug induced:
Vancomycin - most commonly implicated
NSAIDs
Penicillins and other antibiotics
Diclofenac
Lithium
Captopril
Target antigens:
LAD-1 - This is the most frequently identified target and represents the cleaved extracellular portion of BP180
Other antigens that can be targeted - BP 180, BP 230, Type VII Colalgen, Laminin-332
Clinical pearl:
Most patients have both IgA and IgG antibodies and there can be a significant overlap between BP and LABD.
The key distinguishing feature is that IgA flourescence intensity is higher than IgG and C3 on DIF
Clinical presentation
Skin:
Tense blisters, urticated plaques, and erosions
"String of pearls" or "cluster of jewels" sign: annular lesions with peripheral bullae arranged in a rosette pattern (pathognomonic)
Distribution: trunk, limbs, face
Lesions heal WITHOUT scarring
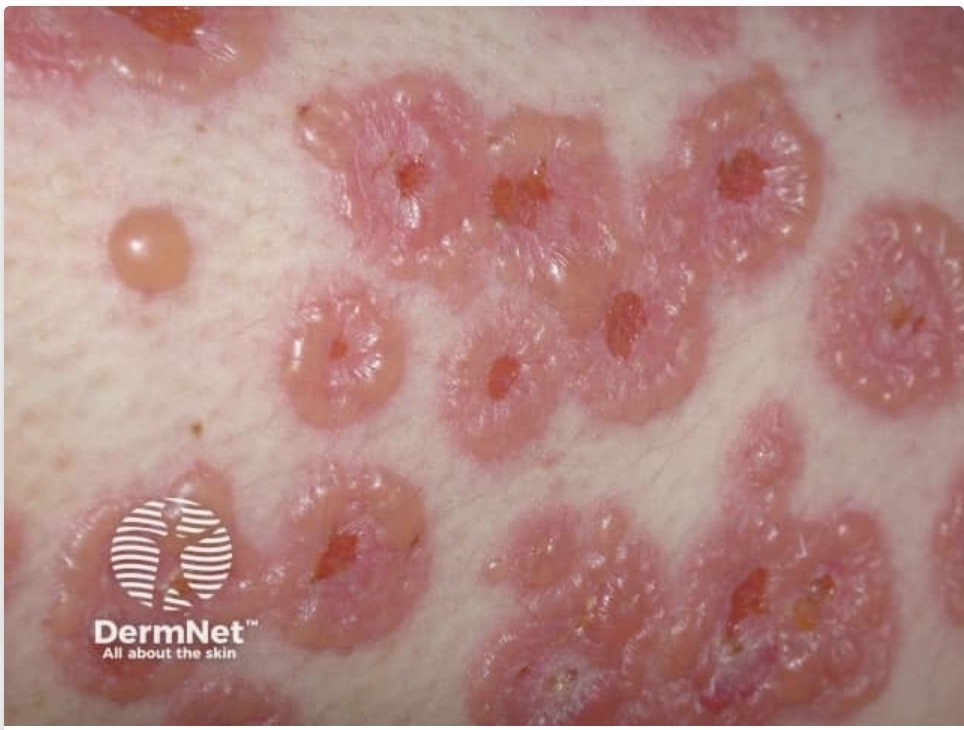
String of pearls sign
https://dermnetnz.org/topics/linear-iga-bullous-disease
Childhood LABD (‘Chronic bullous disease of childhood’)
Perioral and perineal distribution characteristic
Also affects trunk and limbs
Annular configuration with "cluster of jewels" pattern often prominent
Lesions may be very itchy
Mucosal Involvement:
Occurs in up to 70% of patients
Oral mucosa: erosions, can overlap with MMP
Tongue involvement may be prominent (especially IgA-predominant MMP)
Ocular, nasal, pharyngeal, laryngeal involvement possible
May scar at mucosal sites (overlap with MMP)
Investigations
H&E:
Sub-epidermal blister
Inflammatory infiltrate with numerous NEUTROPHILS
Neutrophils arranged at dermal papillae (papillary microabscesses)
May be indistinguishable from DH or BP histologically
Ddx sub-epidermal split with many neutrophils:
Linear IgA
Bullous Lupus
Dermatitis herpetiformis
(still need to consider BP in any sub-epidermal split regardless of infiltrate)
DIF:
LINEAR IgA deposition at the basement membrane zone (defining feature)
May also have IgG and C3, but IgA intensity is HIGHER
This contrasts with pemphigoid where IgG/C3 predominate
Salt-Split Skin:
Can localise to the roof or floor depending on the target antigen
Roof (epidermal side): LAD-1, BP180 targets
Floor (dermal side): type VII collagen, laminin-332 targets
ELISA:
May be positive for BP 180, BP 230 or Type VII Collagen
There is no commercially available ELISA in UK/Ireland for LAD-1
Management:
Linear IgA disease can be very responsive to Dapsone and you can often see dramatic responses to 48-72 hours of Dapsone treatment
Prognosis:
Good prognosis overall
Most children go into remission within 2 years
Drug-induced LABD resolves about 4-8 weeks after stopping the culprit drug
Adult idiopathic disease may be more persistent
EPIDERMOLYSIS BULLOSA ACQUISITA
EBA is a rare, acquired autoimmune sub-epidermal blistering disease caused by IgG autoantibodies to type VII collagen, the main component of anchoring fibrils at the dermal–epidermal junction.
Key points
Onset: 45–55 years
Sex: M = F
Incidence: ~0.2–0.5 per million/year
Associated with inflammatory bowel disease, especially Crohn’s disease
Clincial features
Two main phenotypes (may coexist)
Mechanobullous (classical) variant:
Trauma-induced tense blisters
Dorsal hands, elbows, knees, feet
Healing with scarring and milia (hallmark)
Nail dystrophy or loss
Inflammatory (BP-like) variant:
Widespread tense bullae on erythematous skin
Mimics BP, LABD, or MMP
Scarring less prominent initially
Mucosal disease (common, often severe)
Oral ulcers → scarring, ankyloglossia, microstomia
Oesophageal strictures, dysphagia
Ocular conjunctival scarring
Laryngeal, anogenital involvement
Oral clues
Deep sub-epidermal ulcers
Tongue scarring
Relative gingival sparing (vs MMP)
Investigations
H&E
Sub-epdermal blisters +/- neutrophils seen
Can be ‘cell poor’ with minimal inflammatory infiltrate
DIF:
Linear IgG +/- C3 seen at BMZ
Salt-split skin:
Binds to the floor
Serration pattern:
U-serrated
ELISA:
Type VII collagen
Management
Due to the rarity of the disease can be difficult to know which are the most effective medications for this condition
Mechanobullous variant tends to be quite resistant to standard immunosuppressive medicataions
EBA is often treatment-resistant and usually requires combination therapy.
General care
Minimise trauma
Wound care, infection prevention
Nutritional support if oral disease
Screen for IBD
Treatment considerations:
Topical steroids
Colchicine
Dapsone
Oral corticosteroids
MMF
Refractory disease:
Rituximab
IVIG
Immunoadsorption
Key agents
Colchicine: 500 mcg BD (mechanobullous disease)
Dapsone: useful due to neutrophilic pathology
Prednisolone: ~0.5 mg/kg (usually with steroid-sparing agent)
MMF: preferred steroid-sparing option
Rituximab: refractory disease (slower response than pemphigus)
IVIG: severe disease or contraindication to immunosuppression
Mucosal disease
Treat aggressively
Early ophthalmology, ENT, and GI involvement essential
There are many inherited forms of epidermolysis bullosa
This type is an acquired mechanobullous disorder
Get antibodies to type collagen 7 which constitutes part of the anchoring fibrils which attach the basement membrane to the underlying dermis
Clinical:
Get non-inflammatory tense blisters occur at sites of trauma/friction (hands, wrists, ankles, knees) Can get mucous membrane involvement also
As, split is below the lamina densa it scars with scarring milia and atrophic scarring
May get some mucous membrane invovlement and in some cases can get oesophageal stenosis
H&E:
Sub-epidermal blistering
‘Often cell poor’ (minimal inflammation)
Ddx cell poor sub-epidermal blister:
EBA
PCT
(consiser BP as it is so common)
DIF:
Linear IgG +/- C3 +/- IgA in Basement membrane
Another method that may help differentiate EBA from the pemphigoid diseases is the serration pattern
This involves looking at the linear flourescence and if the overall pattern looks like:
A repeating ‘u’: think Epidermolysis bullosa acquisita or bullous SLE
A repeating ‘n’: think of the other pemphigoid diseases -BP, MMP, anti-p200 pemphigoid, linear IgA dermatosis
IIF with salt split skin test:
Antibodies tend to attach to the dermal side
ELISA:
Positive antibodies against Collagen VII
BULLOUS LUPUS ERYTHEMATOSUS (BSLE)
BSLE results from autoantibodies targeting BMZ antigens, most often type VII collagen
Epidemiology:
Affects < 5% of SLE patients
Strong female predominance (approx 9:1)
Typical onset 20-40 years
More common in patients of African descent
May precede or complicate established SLE
Target antigens:
Type VII Collagen is the most common
BP 180, BP 230, Laminin-332 can also be affected
Clinical features:
Cutaneous:
Widespread tense vesicles and bullae
On erythematous or normal skin
Often sun-exposed areas (face, neck, trunk, arms)
Interestingly despite collagen VII being targeted lesions usually heal without scarring or milia (in contrast to EBA)
May coexist with cutaneous lupus features
https://dermnetnz.org/topics/bullous-systemic-lupus-erythematosusMucosa:
Oral involvement in up to 50%
May affect pharynx, oesophagus, conjunctiva
Typically heals without scarring
Investigations
Lab investigations to include:
FBC, U&E, LFT
ANA
Anti-dsDNA - often positive
Low complement levels may be seen
ENA/antiphospholipids antibodies if indicated
H&E:
Sub-epidermal blister
Prominent neutrophilic infiltrate
May see neutrophilic microabscesses at dermal papillae
May see mucin within the dermis
DIF:
Linear or granular IgG/IgM/IgA/C3 at the DEJ
Salt split skin:
Binds to the floor
Serration:
u-serrated pattern
ELISA:
Type VII collagen usally positive
Management
Supportive care:
Sun protection advice
Treat underlying SLE activity as would normally do
First-line:
Dapsone 25–150 mg daily (start low, titrate)
Rapid response (days–weeks)
Second line:
Systemic corticosteroids
Hydroxychloroquine
MMF
Azathioprine
Rituximab
Prognosis:
Transient in the majority of cases and usually regresses with no further flares, irrespective of the activity of the systemic disease.
Typically resolves without milia or scarring but can leave hypo- or hyper-pigmentation.
DERMATITIS HERPETIFORMIS
Dermatitis herpetiformis is an IgA-mediated sub-epidermal blistering disease and represents the cutaneous manifestation of coeliac disease.
It is defined by granular IgA deposition in the dermal papillae.
Key points
Predominantly affects people of Northern European descent
Typical onset: 30–40 years (can occur at any age)
Male predominance (≈2:1)
Incidence: ~1–10 per 100,000/year
Association with coeliac disease
All patients have gluten-sensitive enteropathy on biopsy
Only 10–20% have overt GI symptoms
Severity of skin disease does not correlate with gut involvement
Conversely, around 15-25% of Coeliac patients will have DH
Clinical pearl:
Diagnosis of DH mandates coeliac work-up and lifelong management due to lymphoma risk.
Pathogenesis:
DH results from a gluten-driven IgA immune response with cross-reactivity between gut and skin transglutaminases.
Target antigens:
Tissue transglutaminase (TG2) – gut (coeliac disease)
Epidermal transglutaminase (TG3) – skin
Mechanism
Gluten exposure in genetically susceptible individuals
IgA antibodies to TG2 form in the gut
Cross-reactive IgA to TG3 develops
IgA–TG3 complexes deposit in dermal papillae
Neutrophil recruitment → protease release
Sub-epidermal blister formation
Clinical features
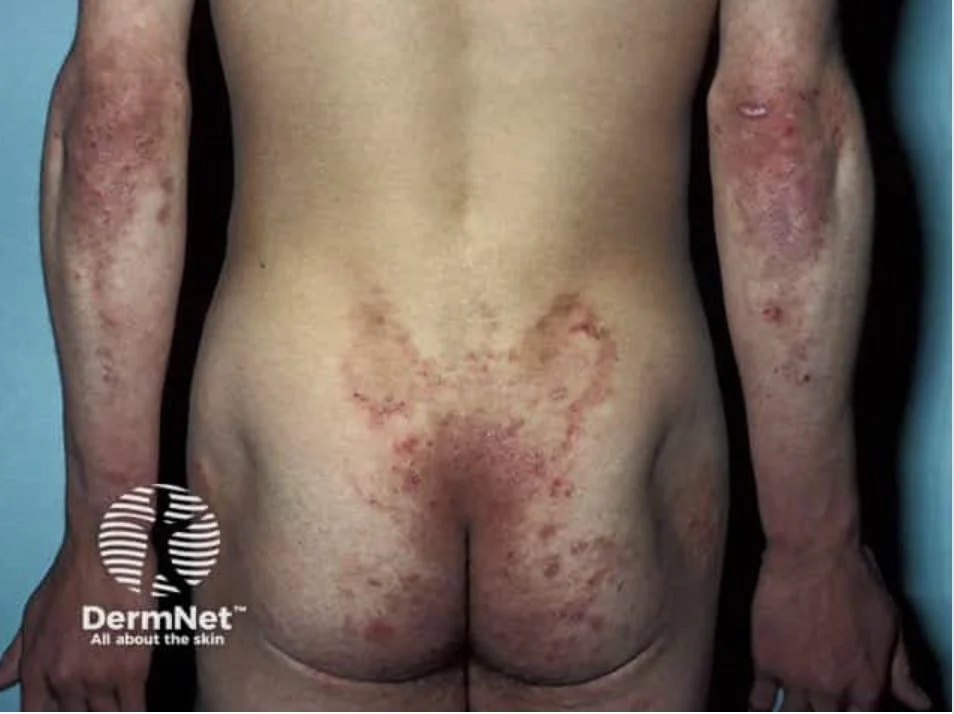
Skin:
Intensely pruritic papules, vesicles, and erosions
Vesicles often absent due to scratching
Grouped (“herpetiform”) and symmetrical distribution
Clinical pearl: Patients usually present with excoriations and not blisters
Classic sites:
Extensor elbows and knees
Buttocks (highly characteristic)
Scalp, scapulae, sacrum
https://dermnetnz.org/topics/dermatitis-herpetiformis
Systemic associations:
GI symptoms uncommon
Iron deficiiency anaaemia
Associated autoimmune disease - thyroid disease, T1DM, Pernicious anaemia
Investigations
H&E:
The most characteristic findings occur at the tips of the dermal papilla
Neutrophilic microbascesses
May see some eosinophils also
‘Shaggy fibrin deposition’
As inflammation progresses you get dermal papillary oedema and sub-epidermal blistering
DIF:
Granular IgA in dermal papillae +/- C3
Serology:
IgA anti-tTG: positive in >90%
IgA anti-endomysial antigen: highly specific
IgA anti-TG3: more specific for DH (research/specialist labs only)
Note: Standard lab tTG assays measure tTG2 (gut) only, not tTG3 – but tTG2 is sufficient for clinical screening.
Check total IgA – ~2% are IgA deficient → use IgG-based tests (tTG-IgG, DGP-IgG)
Gluten-free diet pitfall:
Serology normalises within 6–12 months on a GFD. If already gluten-free:
DIF remains positive longest (months–years) → most reliable test
Gluten challenge: 6–8 weeks before repeating serology (often poorly tolerated)
Always request serology before dietary modification where possible.
Systemic Workup:
FBC, Iron, B12, Folate, TFT
Gastroenterology referral should be considered
Villous biopsy - will show villous atrophy and crypt hyperplasia
Management
Strict lifelong gluten-free diet (GFD)
Treats both skin and gut disease
Reduces lymphoma risk
Skin improvement takes months–years
Dapsone (rapid symptomatic control)
25–50 mg daily → up to 100–200 mg
Dramatic response usually seen within days
If they fail to respond to dapsone reconsider the diagnosis
Alternatives:
Sulfapyridine
Sulfasalizine
Long-term prognosis:
Many patients can discontinue dapsone with strict gluten free diet
Monitor coeliac serology for compliance
Prognosis:
Excellent with adherence to GFD
Increased lymphoma risk with poor dietary compliance
Linear IgA disease
Question 1
Metabolic:
Porphyrias:
Metabolic disorders caused by altered activity of enzymes in the heme biosynthesis pathway
Porphyria can be subdivided into 3 main groups based upon clinical presentation:
Acute intermittent (hepatic) porphyrias - neurological symptoms
Cutaneous porphyria
Acute and cutaneous - neurologic and cutaneous symptoms
Acute hepatic: Acute intermittent porphyria
Acute and cutaneous: Hereditary coproporphyria, variegate porphyria
Cutaneous: PCT, CEP, EPP, Hepatoerytrhopoietic porphyria
Three ways in which porphyrias present in the skin:
Immediate painful photosensitivity:
Erytrhopoeitic porphyria
Mutilating photosensitivity
Congenital erythropoeitic porphyria (Gunter’s disease)
Other rare severe homozyghous porphyrias
Bullous porphyrias - skin fragility and bullae
Porphyria cutanea tarda
Variegate porphyria
What causes Porphyria?

Note:
Linear tetrapyroll also referred to as hydroxymethylbilline
Uroporphyrinogen-1synthase aka porphobilinogen deaminase (PBG-D) or hydromxymehtylbilane synthase
Haem is found in haemoglobin, myoglobin and other haem protens such as cytochromes
Haem is made from the precursors glycine and succinyl CoA
These are converted to haem by 8 chemical reaction steps
Each step is catalysed by a specific enzyme
If one enzyme has reduced activity and is not working properly you will get an acculumation particularly of the biochemical on which the enzyme works
If the biochemical is a porphyrin as in the case of some of the intermediaes above then porphyrins accumulates
Acute porphyrias occur due to direct cytotoxic effect of ALA and PBG on neural tissue
Cutaneous porphyrias due to toxic metabolites poteniated by sunlight (UV causes free radical formation)

AIP: PBG deaminase (aka uroporphyrinogen 1-synthase)
CEP: UPC
PCT: UROD
HC: Coproporhyringoen
VP: PPO
EPP: Feroocheletase
Mechanism of action
Porphyrins can be phototoxic
A photon of visible violet light (approx 408-410nm) is absorbed by porphyrin
This causes the porphyrin to be in an excited state (called a ‘triplet state’)
This can then react with oxygen and turn oxygen into its activated state (‘singlet oxygen’)
The singlet oxygen then directly damages local tissues
So think of porphyrins as little machines that convert visible light photons into tissue damage via singlet oxygen
As mentioned previously porphyrias can present differently in skin eg,
EPP causes an immediate, painful photosensitivity
PCT causes fragility and bullae particularly on back of hands
One hypothesis is this difference is due to the solubility of the different porphyrins due to the number of carboxylate groups (COOH) each porphyrin has which alters the solubility of the porphyrin

Uroporphyrin is water soluble
Protoporphyrin lipid soluble (and not water soluble)
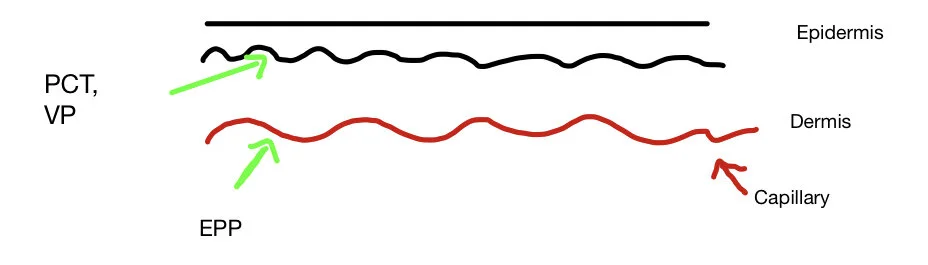
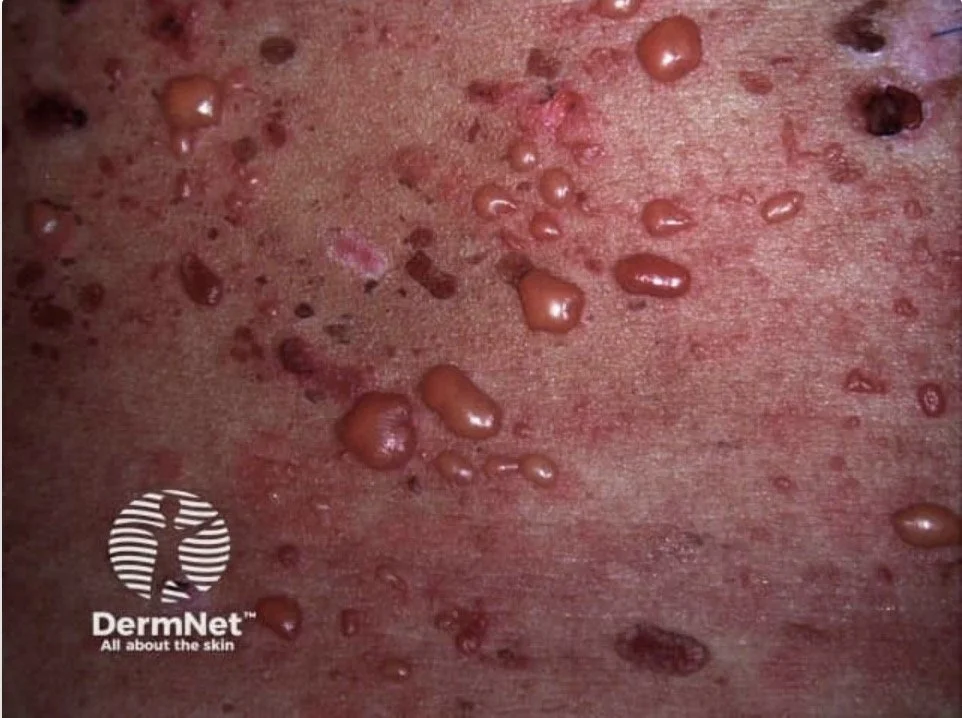
In PCT, uroporphyrin accumulates in the skin and as it is water soluble it can diffuse up to upper dermis
It is then activated by light and the phototoxic reaction occur in the upper dermis causing a split with blistering
In EPP, the protoporphyrin accumulates in the red cells and does not diffuse further than the endothelial lining of small blood vessels.
Light produces endothelial necrosis in the upper dermis causing typical pain and oedema
Porphyria cutanea tarda
PCT is the commonest of the porphyrias
Due to an acquired or inherited deficiency in the activity of the hepatic enzyme uroporphrinogen decarboxylase which usually converts uroporphyringoen III into Coproporphyrinogen III

Decrease in activity of this liver enzyme can be multifactorial
It is due to an inhibitor UROD called uroporphomethene
This is produced particularly in the presence of an oxidising environement and increased iron in the liver

Certain conditions predispose to an environment in which this inhibitor is produced leading to PCT:
Haemochromatosis (20%)
Hepatitis C (15%)
Alcohol (50%)
Oestrogen: eg HRT (50% females)
Smoking
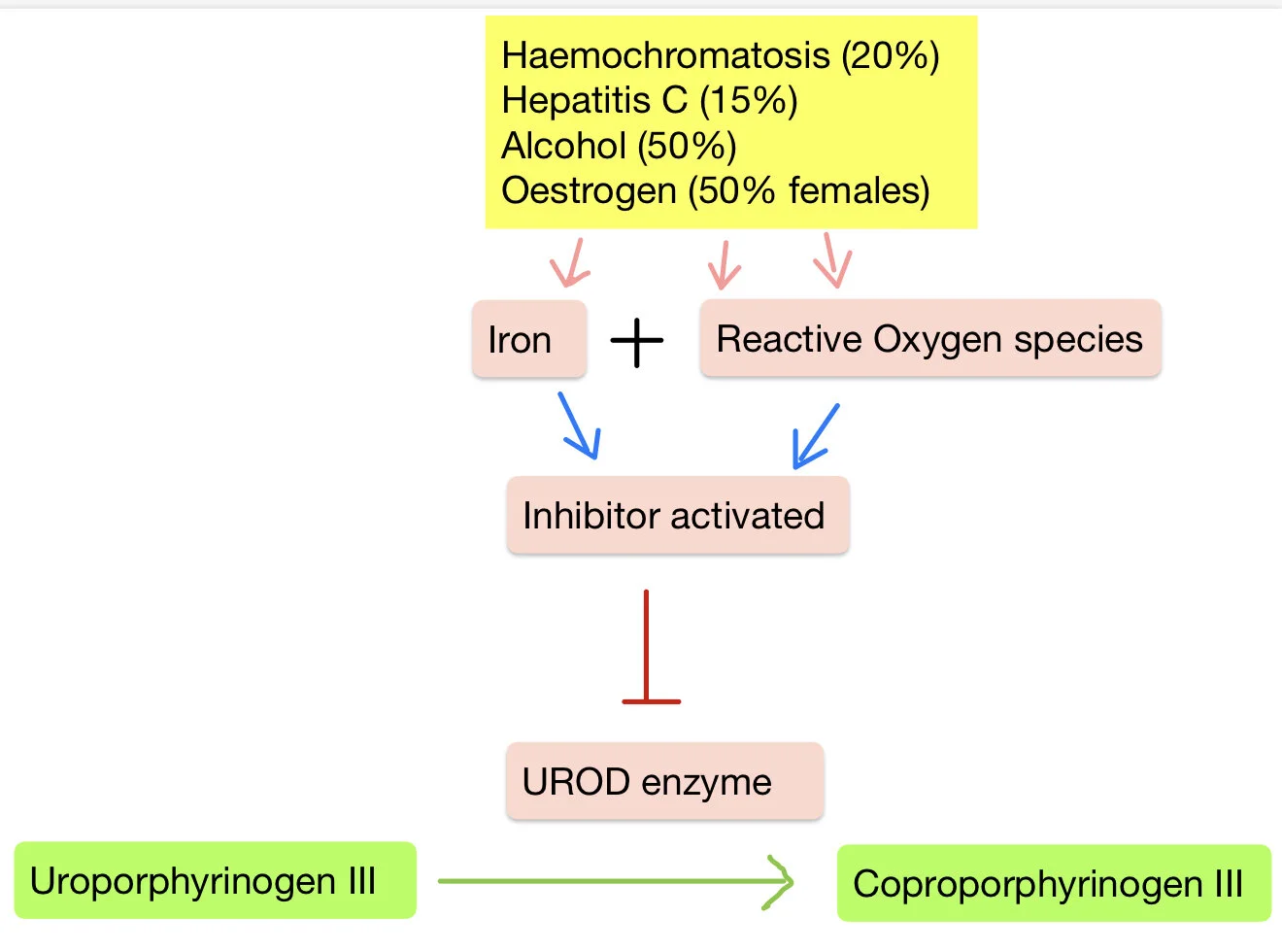
So in summary, inhibition of UROD leads to a build up of uroporphyrinogen III
Genetic factors may be present:
A UROD mutation which is seen in 25% of patients makes the enzyme not as effective
Therefore it requires less inhibitor activation to produce the disease so often get the disease at an earlier stage
Reduced activity of UROD leads to accumulation of porphyrins (eg uroporphyrin) which can be transported to the skin.
These porphyrins are ‘photosensitizing’
-Sunlight (UVA and visible light ranges) cause them to become excited and damage surrounding tissue.
Clinical:
Sun sensitivity
Increasingly fragile skin on back of hands and forearms
Get multiple erosions and blisters
This heals with scarring in the form of milia (tiny cysts)
Can also get hyperpigmentation on face and neck and hypertrichosis (excess hair)
Diagnosis:
Skin biopsy: sub epidermal blistering
All porphyrias that cause skin lesions have elevated plasma porphyrins
Elevated urinary uroporphyrin (and others) and fecal porphyrins indicate PCT
[Should probably do iron studies, HIV and viral hepatitis testing in these patients]
(Urine may look reddish/‘tea’ coloured)
Avoid unnecessary exposure to strong light
Pseudoporphyria
Looks like porphyria cutanea tarda
BUT porphyrin levels are normal
Reaction here is to UV light and not visible light like PCT
Symptoms:
Photosenstivity and fragility
Tense blisters at minor trauma sites (and erosions/scabs)
Often on hands and feet
Heal with scars and milia
Usually medications: (medications react with sunlight to cause photoxic reaction in skin)
· NSAIDs
· Antibiotics-doxycycline
· Diuretics- thiazides, furosemide, bumetanide
· Retinoids- acitretin, isotretinoin
· OCP
Investigations:
Phototesting to confirm photoxic action of the supected agent
Skin biopsy of blister
May check porphyrins in blood, urine and faeces to rule out PCT
Management:
Withdraw offending agent (usually goes away in weeks but cn persist)
Sun protection
Variegate porphyria:
Due to protoporphyrinogen oxidase deficiency
Looks exactly like PCT in skin
Often presents earlier than PCT (teens)
Very important to diagnose as if misdiagnose can prove fatal
Variegate porphyria can cause acute, systemic internal attacks
Acute porphyria signs/symptoms:
AIP, Variegate porphyria, hereditary coproporphyria
Can present with sudden life-threatening crisis
Most patients have one or only a few attacks followed by resolution of syndrome
Attacks occur rarely in childhood with incidence increasing after puberty
Manifest by neurovisceral symptoms and autonomic sequelae
Neuro: fatigue, confusion, seizures, motor neuropathy
GI: abdominal pain, N/V, constipation
Autonomic: hypertensive urgency, fever
Hyponatraemia
(Think in someone with unexplained neurological symptoms in someone with autonomic and/or GI symptoms and hyponatreaemia)