
A lymphoma is a cancer of the lymphocyte and lymphocytes come in three main
sub-types:
T cells
B cells
Natural killer (NK) cells
Any of these can become malignant, though in the skin the overwhelming majority are T-cell (chiefly mycosis fungoides) or, less often, B-cell.
NK-cell lymphomas of the skin are very rare
The majority of systemic non-Hogdkin’s lymphomas are B cell lymphomas whereas the majority of primary cutaneous lymphomas are T cell lymphoma
If have a lymphoma on the skin you want to think:
Is this a primary cutaneous lymphoma?
or
Is it a systemic lymphoma that has spread to the skin (secondary cutaneous lymphoma)?
Primary cutaneous T cell lymphomas
Primary cutaneous T cell lymphomas account for ≈ 80% of primary cutaneous lymphomas
We will discuss B cell lymphoma and blastic primary dendritic cell neoplasm later
Primary cutaneous T cell lymphoma can be classified as follows:
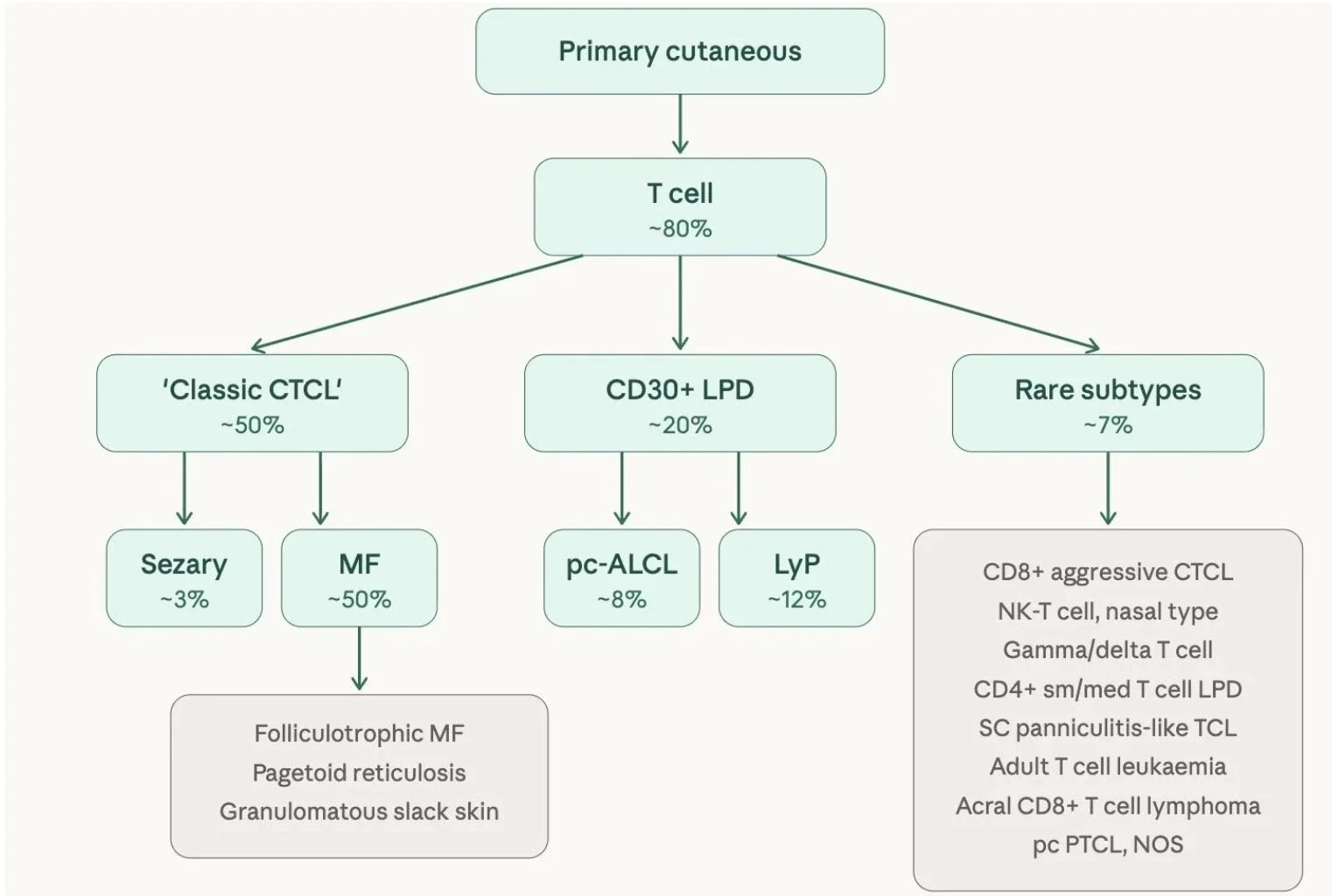
'Classic' CTCL
This is the largest group and includes mycosis fungoides (the commonest cutaneous lymphoma overall, accounting for ~50% of all primary cutaneous lymphomas), its variants (folliculotrophic MF, pagetoid reticulosis, granulomatous slack skin), and Sézary syndrome (~3%)
CD30+ lymphoproliferative disorders (~20%)
Defined by the expression of CD30 on the cell surface
This subgroup is staged and treated differently to MF and includes primary cutaneous anaplastic large cell lymphoma (pc-ALCL) and lymphomatoid papulosis (LyP)
Rare subtypes (~7%) — a collection of less common entities including:
CD8+ aggressive epidermotropic T cell lymphoma
NK-T cell lymphoma, nasal type
Gamma/delta T cell lymphoma
CD4+ small/medium T cell lymphoproliferative disorder
Subcutaneous panniculitis-like T cell lymphoma
Adult T cell leukaemia/lymphoma
Acral CD8+ T cell lymphoma (provisional entity)
Primary cutaneous peripheral T cell lymphoma, NOS
This distinction matters clinically because each group has a different prognosis and management approach. Classic CTCL and CD30+ LPD tend to behave more indolently, whereas several of the rare subtypes can be aggressive
DIAGNOSTIC TOOLKIT
Before diving into individual entities, it helps to be aware of the diagnostic toolkit we have for diagnosing a cutaneous lymphoma. The diagnosis rests on integrating the following:
Clinical picture
Histology (H&E)
Immunohistochemistry
Molecular clonality studies
± Flow cytometry (if blood involvement queried)
H&E:
Standard H&E staining is the first line and can identify both atypical cells and abnormal patterns of lymphocyte infiltration in the skin.
For example the classic patterns we can see in Mycosis Fungoides are as follows:
Atypical lymphocytes with hyperchromatic, cerebriform nuclei (the convoluted nuclear membrane resembles the surface of the brain).
Epidermotropism — the tendency of these atypical T cells to migrate up into the epidermis. This is a hallmark feature of MF and helps distinguish it from inflammatory mimics, where lymphocytes are usually confined to the dermis. Importantly, the epidermis itself is typically not spongiotic, despite being infiltrated — a useful clue.
Pautrier microabscesses — small intraepidermal collections of these atypical lymphocytes. They are highly specific for MF but only seen in a minority of cases (around 10–20%), so their absence does not exclude the diagnosis.
Immunohistochemistry:
We identify different lymphocytes by the CD markers (proteins) on their surface — think of these as flags. They can be identified by using immunohistochemical stains (antibodies which attach to the marker and stain brown)
The presence or absence of certain proteins helps us decide what types of cells are present and if they are normal or abnormal
For instance:
CD3 marks T cells
CD20 marks B cells
T cells can be subdivided into CD4+ helper cells, CD8+ cytotoxic cells, and regulatory T cells
T cells also normally express a set of pan-T cell markers including:
CD2
CD3
CD5
CD7
— meaning these are present on most mature T cells. However, when a T cell becomes malignant, it can lose expression of one or more of these markers.
Surface receptors and clonality
Lymphocytes also carry unique surface receptors
T cells have a T cell receptor (TCR) — this is the part of the T cell that binds to antigens and activates the immune system.
Every T cell has a slightly different receptor, which is how the immune system can recognise the huge variety of antigens it encounters.
In normal tissue you see a diverse mix of these receptors.
When one cell multiplies out of control, you find clones — many cells with identical receptors
T-Cell Receptor (TCR) Gene Rearrangement Testing
Also known as TCR PCR or high-throughput sequencing, this test analyzes DNA from tissue lymphocytes (e.g., blood, skin, or bone marrow) to detect if a specific T-cell clone is multiplying out of control.
Normal Results (Polyclonal): Healthy tissue contains highly diverse T-cells. On a test, this diversity appears as a smooth, normally distributed waveform.
Abnormal Results (Monoclonal): If a single clone starts to multiply abnormally, a distinct "monoclonal peak" emerges while background diversity drops. As the clone expands, the peak becomes more dominant, which supports a diagnosis of lymphoma.
Important Interpretation Caveats:
Not all clones are cancer: While all cancers are clonal, a monoclonal peak can also be caused by benign reactive processes (like infections, drug reactions, or eczema). In these cases, the clone typically disappears once the underlying trigger resolves.
False negatives: A clone can sometimes be missed if the disease is in its very early stages, or if the tissue sample is too small or degraded.
Flow cytometry
Is a way of counting and sorting lymphocytes based on the CD markers they express on their surface. In CTCL it is mainly used on blood to look for circulating malignant cells.
The sample is put into suspension and individual cells are passed through a laser one at a time. Each cell is tagged with multiple fluorescent antibodies of different colours that target different CD markers, so multiple markers can be detected simultaneously on each cell in a single run — you get CD4 counts, CD8 counts, CD4:CD8 ratio, and marker loss information all at once.
This is useful because normal T cells express a predictable set of surface proteins such as CD7, and malignant T cells often lose one or more of these. Flow cytometry detects abnormal cells by identifying what's missing.
Key findings in CTCL with blood involvement (ie Sézary syndrome or erythrodermic MF) include an increased CD4:CD8 ratio (≥10), loss of CD7 and/or CD26 on CD4+ cells, and an elevated Sézary cell count
Putting it all together
Going forward, when working up a suspected cutaneous lymphoma, we rarely rely on a single test
The diagnosis is built up from a combination of:
H&E — to look at the morphology of the cells and the pattern of infiltration (eg epidermotropism, cerebriform nuclei, Pautrier microabscesses)
Immunohistochemistry — to phenotype the infiltrate and look for loss of normal T cell markers
TCR gene rearrangement studies — to demonstrate clonality
Flow cytometry — added in when we are thinking about blood involvement (eg suspected Sézary syndrome or erythrodermic MF)
No single one of these is diagnostic on its own — clones can occur in reactive conditions, marker loss can be partial, and morphology can be subtle, especially in early disease
It is the clinicopathological correlation of all the pieces together — alongside the clinical picture — that gets you to the diagnosis
Mycosis fungoides
Is the most common form of CTCL and accounts for 50% of all primary cutaneous lymphomas
Clinical presentation
Typically affects older adults (median age at diagnosis: 55-60 years)
Can occur in children (including a hypopigmented variant)
Usually follows an indolent course with slow progression over years or even decades
General progression: patch → plaque → tumour
Initial lesions have a predilection for the buttocks and other sun-protected areas (“bathing suit distribution”)
In later stages, lymph nodes and visceral organs may become involved
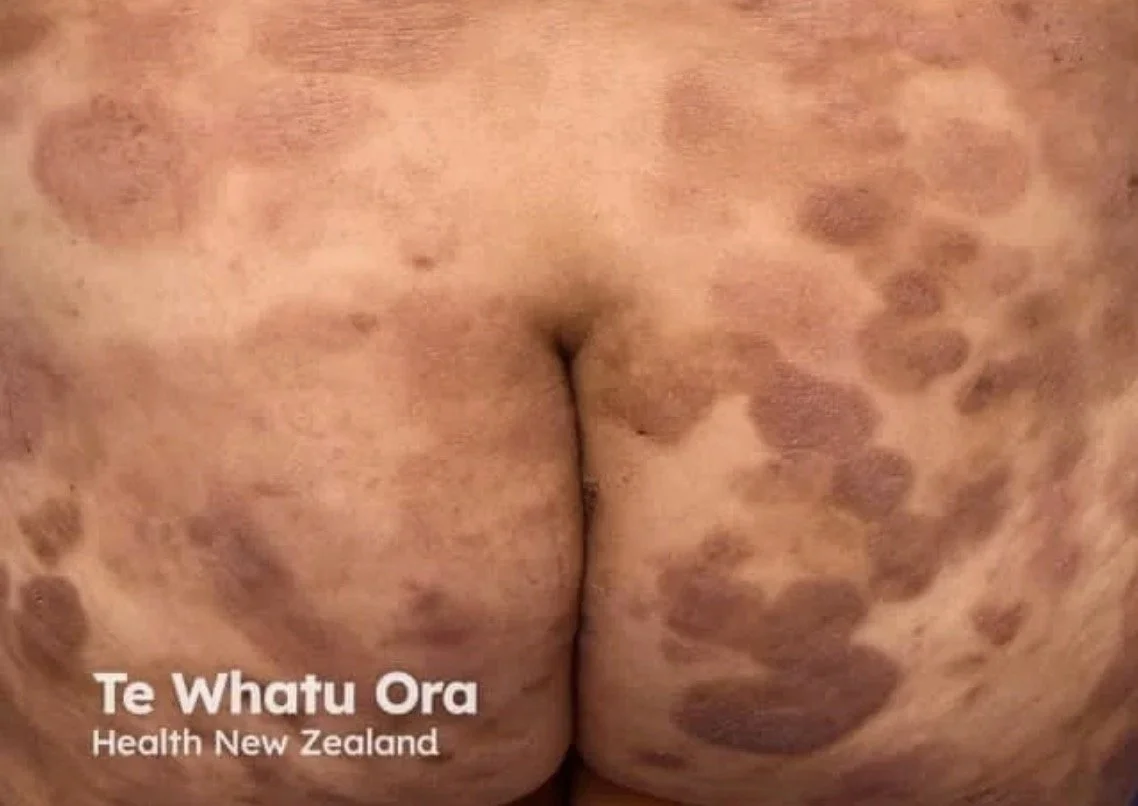
Patch stage:
Flat, erythematous lesions with fine scale
Surface atrophy with fine wrinkling may be seen
Often asymmetrical
Irregular “geometric” shaped lesions
Plaque stage:
Raised, well-demarcated, often violaceous plaques
Thicker and more infiltrated than patches
May co-exist with patches
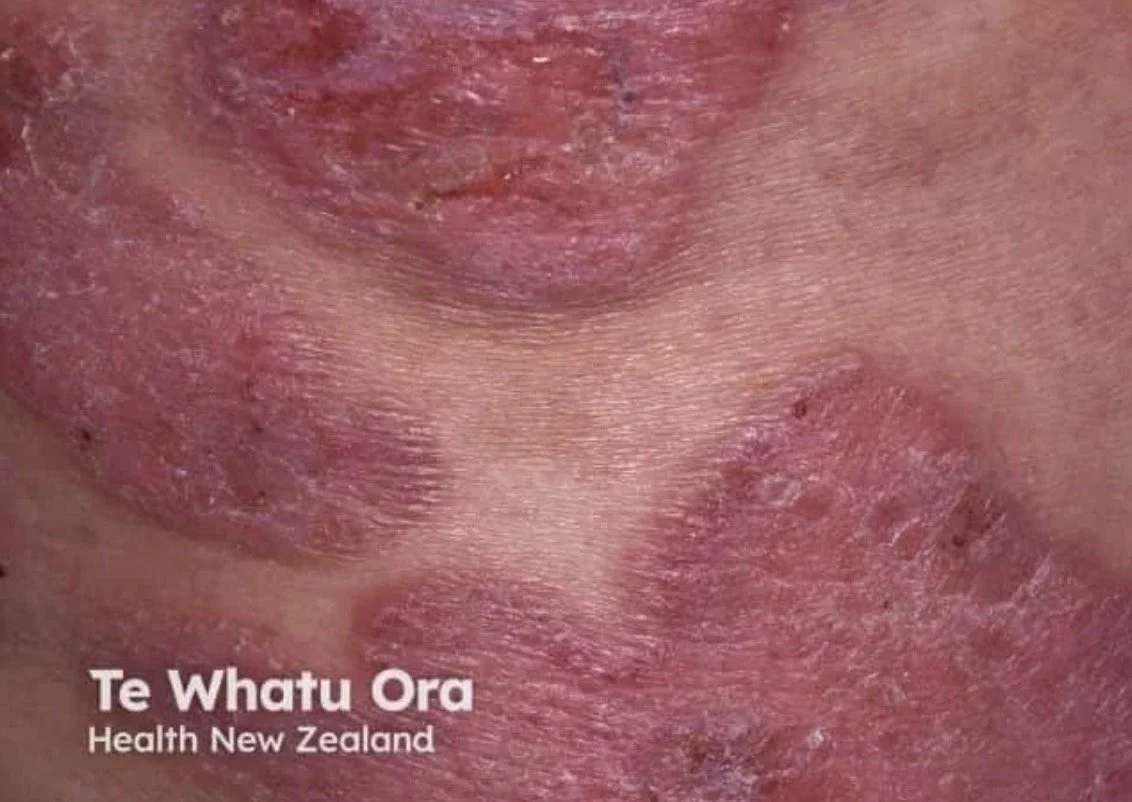
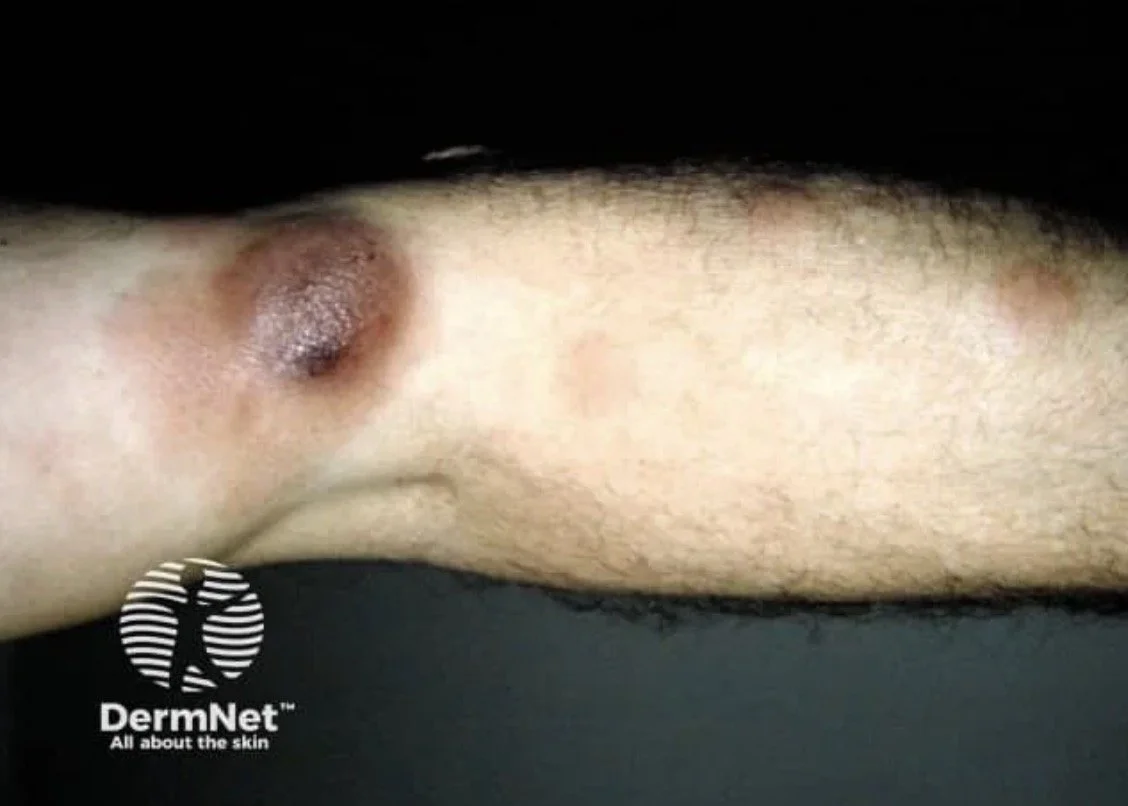
Tumour stage:
Dome-shaped nodules or tumours (≥1 cm)
May ulcerate
Can arise on a background of pre-existing patches and plaques — always look for coexisting patches/plaques
Transformation to a diffuse large cell lymphoma (CD30+ve or −ve) can occur and carries a poor prognosis
Distribution:
Pelvic girdle and shoulder girdle; sun-protected sites
Asymmetrical distribution is characteristic
MF Histopathology
Early patch:
Superficial bandlike infiltrates consisting of lymphocytes and histiocytes
Can get atypical cells with small to medium sized, highly indented (cerebriform), hyperchromatic nuclei
These atypical cells are mostly confined to epidermis (epidermotropism) and may initial be seen around basal layer
Patch:
Superficial band-like infiltrate of lymphocytes and histiocytes are seen in the upper dermis
Atypical lymphocytes — a few small to medium-sized cells with cerebriform, hyperchromatic nuclei (convoluted, looks like a raisin) infiltrate into the epidermis (epidermotropism) and importantly the epidermis is not spongiotic despite being infiltrated (a useful clue to MF)
Classically, the atypical cells are intially found in the basal layer and appear single "haloed" cells (lymphocyte surrounded by small clear vacuole) which then can begin to ‘line up’ up along the dermo-epidermal junction
Pautrier’ microabscesses: Small clusters of atypical lymphocytes within the epidermis. These are pathognomonic but are actually less common in the patch stage than in the plaque stage.
Plaque:
As the disease progresses to a plaque, the cellular infiltrate becomes more "heavy" and the architectural distortion is more obvious.
The epidermotropism becomes more pronounced
May see more Pautrier microabscesses
A denser dermal infiltrate will be seen
More obvious epidermotropism and denser dermal infiltrate
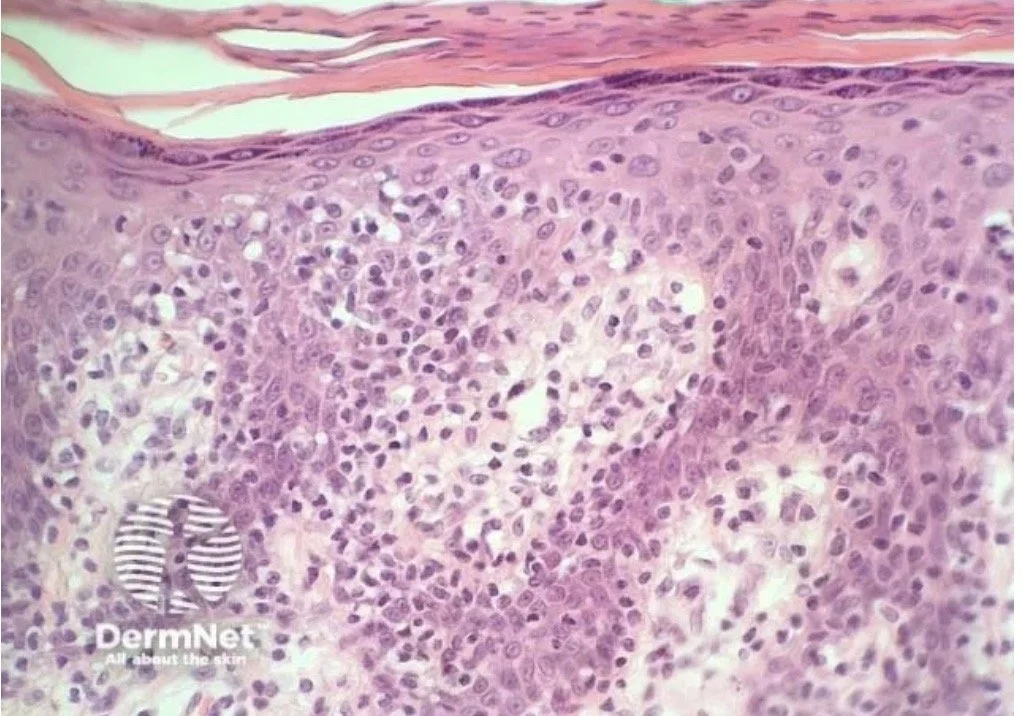
Tumour:
The dermal infiltrate becomes more diffuse and epidermotropism may be lost
The tumour cells increase in size and numbers
Cells can become more pleomorphic
Transformation to a diffuse large cell lymphoma (CD 30 +ve or -ve) can occur and has a poor prognosis
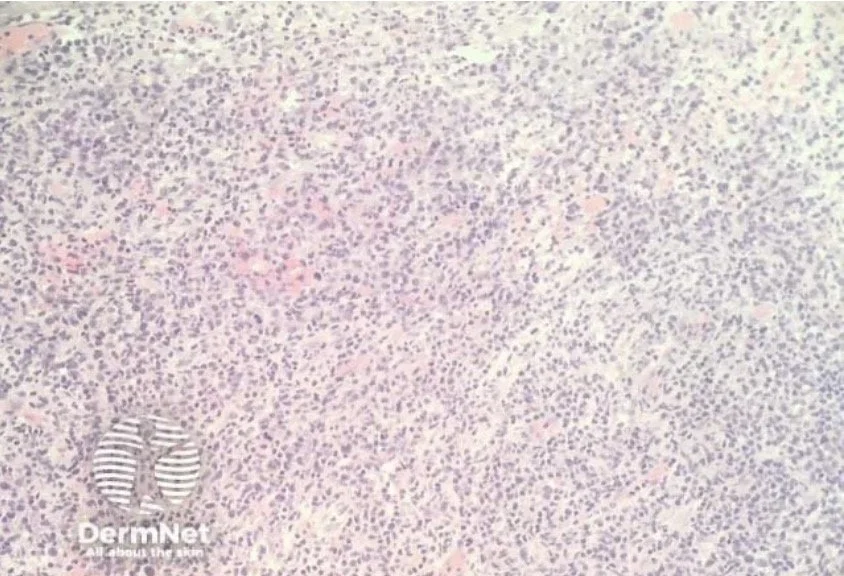
Dense dermal infiltrate in tumour stage
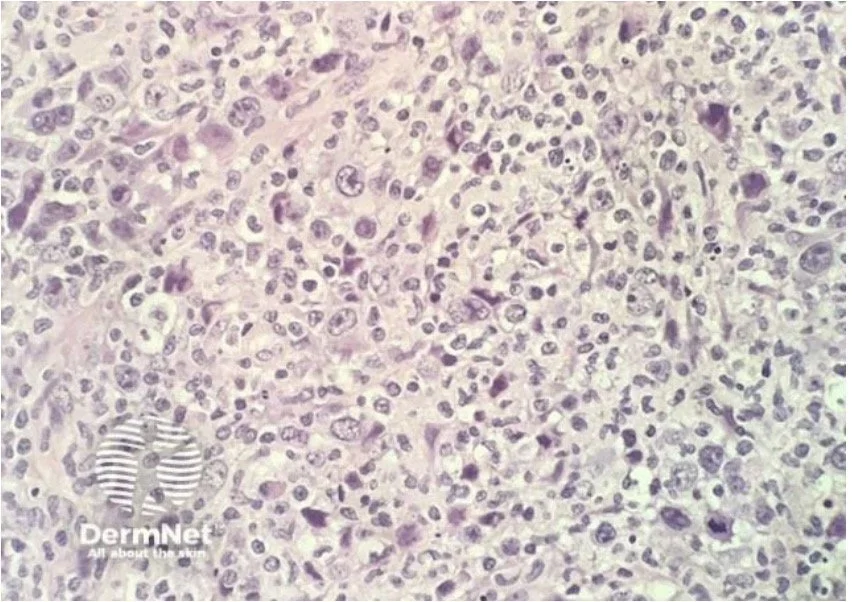
Large cell transformation - poor prognosis
MF Immunohistochemistry
CD3+, CD4+, CD45RO +ve (CD8 -ve usually)
Rarely can be CD4- and CD8+ (particularly in hypopigmented disease)
Can lose pan-T cell antigens (CD2, CD3 and CD5) which can aid in diagnosis
MF Clonality
Clonal T-cell receptor gene arrangement is detected in most cases of MF
Tips regarding TCR gene rearrangement in MF:
The detection of a T cell clone in skin in the absence of consistent histology is NOT diagnostic of CTCL
The presence of an identical T cell clone in multiple skin biopsies (± blood) is useful in confirming a diagnosis of CTCL in patients with consistent histology and clinical features
In patients where diagnosis is uncertain, identification of identical clones from 2 separate skin biopsies helps to confirm CTCL
DNA from different biopsies and different tissues should always be analysed together to provide informative results
T cell clones in the blood alone (especially in older patients) can be incidental and are not necessarily diagnostic
MF Variants
Folliculotrophic MF
This is a variant of MF characterised by a folliculotrophic infiltrate, often with sparing of the epidermis. It frequently affects the head and neck. Most cases (but not all) show mucinous degeneration of the hair follicles (follicular mucinosis). Because infiltrates are associated with the follicles and therefore deeper, skin-directed therapies are often less effective.
Clinical:
Mostly seen in adults (can affect children)
Males > Females
Can present as grouped follicular papules, acneiform lesions, or indurated plaques/tumours
Often associated with alopecia and release of clear mucin from affected areas (mucinorrhoea)
Can be very itchy — progressing pruritus may indicate disease progression
Histopathology:
Perivascular and periadnexal infiltrates with variable infiltration of follicular epithelium by small, medium, and sometimes large hyperchromatic cells with cerebriform nuclei
Epidermis tends to be spared
Alcian blue stains may demonstrate mucinous degeneration of follicular epithelium (“follicular mucinosis”)
May see eosinophils and plasma cells
If eccrine glands are prominently affected it is called syringotropic MF
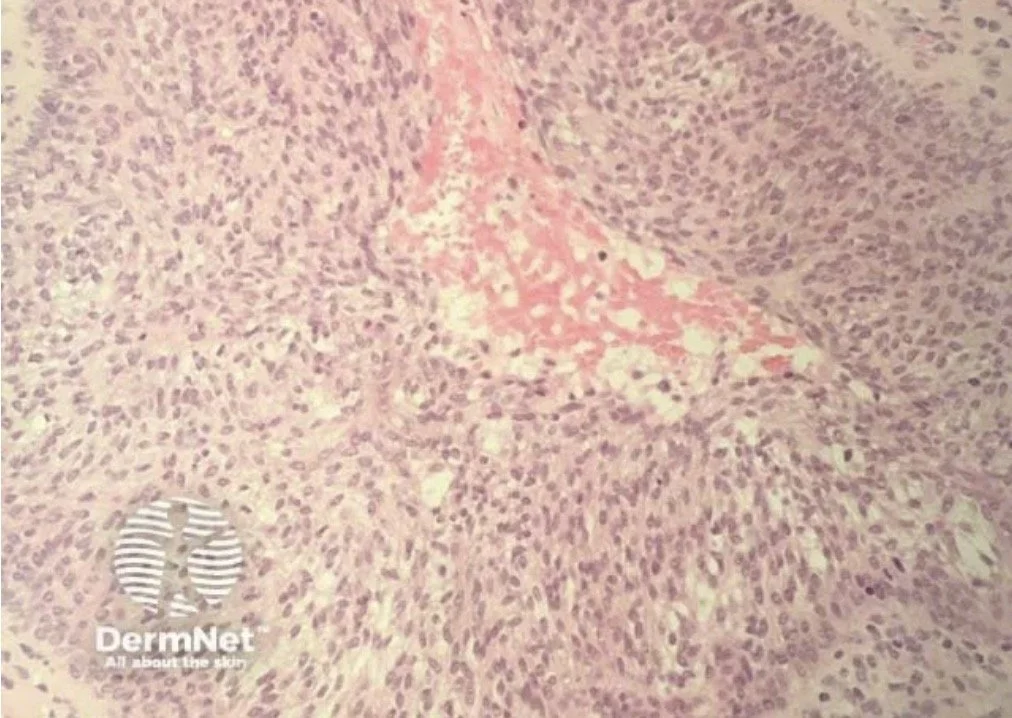
Folliculotrophic MF
This is a variant of MF characterised by a folliculotrophic infiltrate, often with sparing of the epidermis. It frequently affects the head and neck. Most cases (but not all) show mucinous degeneration of the hair follicles (follicular mucinosis). Because infiltrates are associated with the follicles and therefore deeper, skin-directed therapies are often less effective.
Clinical:
Mostly seen in adults (can affect children)
Males > Females
Can present as grouped follicular papules, acneiform lesions, or indurated plaques/tumours
Often associated with alopecia and release of clear mucin from affected areas (mucinorrhoea)
Can be very itchy — progressing pruritus may indicate disease progression
Histopathology:
Perivascular and periadnexal infiltrates with variable infiltration of follicular epithelium by small, medium, and sometimes large hyperchromatic cells with cerebriform nuclei
Epidermis tends to be spared
Alcian blue stains may demonstrate mucinous degeneration of follicular epithelium (“follicular mucinosis”)
May see eosinophils and plasma cells
If eccrine glands are prominently affected it is called syringotropic MF
Pagetoid reticulosis
Pagetoid reticulosis
A rare, indolent variant of mycosis fungoides
Typically a solitary, slowly progressive hyperkeratotic or psoriasiform plaque on a distal extremity, with striking epidermotropism on histology and an excellent prognosis.

Granulomatous slack skin
Granulomatous slack skin
A very rare variant of mycosis fungoides (MF)
Characterised by the slow development of folds or lax skin in the major skin folds.

Sezary Syndrome
Sézary syndrome (SS) is the leukaemic variant of CTCL, defined by a classic triad:
Erythroderma — generalised erythema involving ≥ 80% of body surface area (BSA)
Generalised lymphadenopathy
Sézary cells — neoplastic T cells with cerebriform (brain-like) nuclei, found in skin, lymph nodes and peripheral blood
It is rare — accounting for roughly 3% of all CTCL — and essentially exclusive to adults, with most patients diagnosed in their sixties.
There is a clear male predominance (≈2:1)
Clinical features
Erythroderma, often with marked exfoliation, oedema, lichenification and intense pruritus
Commonly associated with lymphadenopathy, alopecia, onychodystrophy (nail dystrophy) and palmoplantar hyperkeratosis
Clinical tip: The erythrodermic patient can be hard to diagnose. Look for palmoplantar keratoderma and nail dystrophy, and ask about the skin before the erythroderma developed (eg history of patches/plaques in bathing suit distribution)
Making the diagnosis:
Clinical suspicion requires the full triad above: erythroderma (≥80% BSA), peripheral lymphadenopathy, and circulating Sézary cells.
Haematologic (B2) criteria:
To confirm blood involvement and separate SS from erythrodermic MF, two things are required: a clonal blood TCR rearrangement matching the skin clone, plus evidence of high tumour burden in the blood.
The current EORTC/ISCL/USCLC consensus (2022) sets the tumour-burden threshold by flow cytometry — ≥1000/µL clonal CD4+ T cells with an aberrant phenotype (CD4+CD7− or CD4+CD26−).
What changed:
The earlier 2007 ISCL definition also accepted a CD4:CD8 ratio ≥10, or percentage-based aberrant phenotypes (≥40% CD4+CD7− or ≥30% CD4+CD26−). These were dropped in 2022 because they can reflect skewed CD populations rather than true tumour burden. Manual cerebriform Sézary cell counts on a blood film are likewise no longer preferred, owing to poor inter-observer reproducibility.
Ongoing debate:
The 2022 criteria are more specific for SS, but a single-institution study (Chrisman et al., Blood 2024) suggests they may miss a small subset of patients with low blood burden who nonetheless carry a poor prognosis — so this remains an active area of debate.
Skin biopsy:
Findings overlap with MF — a band-like (lichenoid) infiltrate of small-to-medium atypical lymphocytes with cerebriform nuclei in the upper dermis, with epidermotropism and occasional Pautrier microabscesses.
However, the infiltrate and epidermotropism are often much sparser than in MF, so biopsies are frequently non-specific — up to a third are non-diagnostic, and multiple biopsies with clinicopathological correlation are often needed.
Immunohistochemistry typically shows a CD3+ CD4+ CD8− CD45RO+ memory T-cell phenotype, with frequent loss of CD7 and CD26.
Lymph node biopsy:
Excisional biopsy of the largest palpable node is preferred over fine-needle aspiration, which cannot show nodal architecture. Finding the same T-cell clone in node and skin supports the diagnosis of SS.
Bone marrow:
Not routinely required for diagnosis or staging — marrow involvement is uncommon and rarely alters management. Reserve it for unexplained cytopenias or suspected large-cell transformation or a second haematological malignancy.
Making a diagnosis - MF/SS
Making the diagnosis is the key first step in managing MF/SS, and it often takes time to secure.
CTCL is notorious for mimicking benign inflammatory dermatoses, and the diagnostic work usually starts with history and skin examination, paying close attention to clues that point in the right direction, for example:
Patches in sun-protected sites (the "bathing-trunk" distribution) evolving over years through patches → plaques → tumours (MF)
Or an erythrodermic patient with alopecia, nail dystrophy, ectropion, and marked lymphadenopathy (SS)
Repeat biopsies are often needed — early MF in particular is histologically subtle, and a single non-diagnostic biopsy does not exclude the disease.
Studies that help confirm CTCL on tissue include H&E histopathology, immunohistochemistry (T-cell markers and aberrant loss of CD7/CD26), and TCR gene rearrangement studies to demonstrate clonality.
Where appropriate — particularly with erythroderma, suspected SS, or advanced MF — the workup extends beyond the skin: peripheral blood film, flow cytometry, and blood TCR rearrangement to assess for blood involvement, and excisional biopsy (favoured over FNA) of any palpable lymph node to look for nodal disease and matching clonality.
All patients with suspected CTCL should have a standard panel of blood tests at first assessment. These help establish a baseline, exclude important differentials, and flag features that will influence the depth of staging required.
Baseline invetigations suspected CTCL
Standard panel for all patients with suspected CTCL
Haematology
Biochemistry
Serology & immunology
MF/SS Staging
Once the diagnosis of MF/SS is made, staging defines the extent of disease which helps define prognosis and guide treatment.
The system used worldwide is the ISCL/EORTC TNMB classification, first published by Olsen et al. in 2007 and updated in 2022. MF/SS is staged differently from all other cutaneous lymphomas
It assesses four compartments:
T — skin
N — lymph nodes
M — viscera
B — blood
The depth of staging investigations should match the disease. A patient with a few patches needs little more than clinical examination and baseline bloods. A patient with erythroderma, lymphadenopathy, or tumours needs imaging, lymph node biopsy, and flow cytometry.
Skin (T)
Skin involvement depends on lesion type (patch, plaque, tumour, erythroderma) and body surface area (BSA).
A useful rule of thumb at the bedside: the patient's palm plus all five fingers ≈ 1% BSA
T1 and T2 can be sub-stratified into a (patch only) or b (plaque ± patch). This does not change stage but carries prognostic weight — patients with plaques have a worse outcome than those with patches alone
Skin involvement
Clinical tip: A single nodule >1 cm in a patient with otherwise widespread plaques moves them from T2 to T3 and from early to advanced stage — a major prognostic and management shift
The modified Severity-Weighted Assessment Tool (mSWAT) quantifies skin tumour burden by weighting lesion type (patch ×1, plaque ×2, tumour ×4) against regional BSA. Score ranges 0–400, and it is the standard tool for monitoring response
Lymph Nodes:
Should assess for any lymph nodes clinically
Any peripheral node >1.5 cm, or firm/irregular/fixed/clustered, is considered clinically abnormal and warrants further evaluation
CT neck/chest/abdomen/pelvis (with contrast) is indicated for :
T3 (tumour-stage)
T4 (erythroderma)
Palpable peripheral lymphadenopathy
B-symptoms
Abnormal bloods (raised LDH, cytopenias)
It can also be consider in T2b disease (patches/plaques ≥ 10% BSA)
Lymph node involvement
Clinical tip - FNA is inadequate:
Nodal architecture must be assessed for staging
Excisional or core biopsy is the gold standard, taken from the most clinically or PET-avid node
Both H&E and TCR clonality should be requested
Visceral involvement:
Visceral involvement
Common sites for visceral spread are lung, liver, spleen and CNS.
Bone marrow involvement was formally defined within visceral evaluation in the 2022 ISCL update, and bone marrow biopsy is recommended where there is unexplained cytopaenia or where systemic involvement would change management
Blood (B):
Blood classification is what distinguishes Sézary syndrome from erythrodermic MF, and its definition has been refined twice since 2007
The 2022 ISCL/USCLC/EORTC update (Olsen, Blood 2022) is the current standard
Flow cytometry has replaced morphological Sézary cell counting — manual counts are subjective and poorly reproducible.
Blood involvement is measured by counting the abnormal (clonal) Sézary cells circulating per microlitre. These cells are identified on flow cytometry by their loss of normal T-cell markers — typically CD4+ cells that have lost CD7 or CD26
Each B level can be further sub-classified a (no clone, or clone not matching skin) or b (clone matching skin)
B0b confers a worse prognosis than B0a in early-stage disease, even though both are technically B0
Pearl: B classification has limited prognostic relevance in patch/plaque (T1/T2) disease — most early-stage patients are B0, and there is no clear survival difference between B0 and B1. Some guidelines therefore consider flow cytometry optional in T1/T2
It is essential in T3, T4 and any erythrodermic patient.
Blood involvement
Putting it all toghether:
The four TNMB classifications combine to give a clinical stage from IA to IVB.
MF / Sézary Syndrome — Overall Stage Grouping
ISCL/EORTC TNMB stage groupings with defining clinical features.
| Stage | T | N | M | B | Defining feature |
|---|---|---|---|---|---|
| IA | 1 | 0 | 0 | 0–1 | Limited patches/plaques (<10% BSA) |
| IB | 2 | 0 | 0 | 0–1 | Generalised patches/plaques (≥10% BSA) |
| IIA | 1–2 | 1–2 | 0 | 0–1 | Patches/plaques + clinically abnormal nodes |
| IIB | 3 | 0–2 | 0 | 0–1 | One or more tumours |
| IIIA | 4 | 0–2 | 0 | 0 | Erythroderma, no/minimal blood involvement |
| IIIB | 4 | 0–2 | 0 | 1 | Erythroderma, low-level blood involvement |
| IVA1 | 1–4 | 0–2 | 0 | 2 | High-level blood involvement (Sézary range) |
| IVA2 | 1–4 | 3 | 0 | 0–2 | Effaced lymph nodes |
| IVB | 1–4 | 0–3 | 1 | 0–2 | Visceral involvement |
Early vs advanced:
IA, IB and IIA are early-stage and managed with skin-directed therapy
Note: IA or IB can have a small amount of blood involvement (B0,B1)
IIB onwards is advanced. The transition point is the appearance of a tumour, erythroderma, effaced nodes, B2 blood, or visceral disease.
Sézary syndrome: SS is, by definition, stage IVA1 or higher (T4 + B2). It is distinguished from erythrodermic MF (T4 with B0/B1) by the level of blood involvement.
Additional investigations for advanced disease
Further workup when advanced disease is suspected
Prognosis in MF/SS
The prognosis in MF/SS is historically determined by TNMB stage, but stage alone does not capture all of the clinically important risk.
For instance early-stage MF usually has a favourable prognosis, particularly patch-only disease
However, some patients with early-stage disease have higher-risk features such as plaques, folliculotropic disease, abnormal nodes, older age or large-cell transformation. So two patients with the same stage can behave very differently
Also advanced-stage disease prognosis does not progress neatly by stage number. For example, stage IIB tumour-stage MF generally has a worse prognosis than stage IIIA erythrodermic MF without significant blood involvement
This is why prognosis is best thought of in two layers:
TNMB stage — how much skin, node, blood and visceral disease is present
Additional prognostic factors — clinical and pathological features that refine risk within each stage
Pearl:
Stage tells you where the disease is
Prognostic factors tell you how the disease is behaving
There are two main prognostic indices to help refine risk within stages:
CLIPi (Benton, EJC 2013) — retrospective analysis of 1502 MF/SS patients from the UK St John's cohort, identifying adverse prognostic factors for early- and advanced-stage disease
PROCLIPI (Scarisbrick, Blood 2025) — prospective international registry of >2000 patients across 46 expert centres, refining and validating the prognostic model for advanced disease
For early-stage disease (IA–IIA), the working model at time of writing (March 2026) remains CLIPi
PROCLIPI is still validating a prospective early-stage successor
Five adverse factors at diagnosis:
Male sex
Age >60
Plaques (T1b/T2b rather than patch only)
Folliculotropic disease
N1 or Nx
| Risk group | Factors | 10-year OS |
|---|---|---|
| Low | 0–1 | 90% |
| Intermediate | 2 | 76% |
| High | 3–5 | 49% |
In October 2025, PROCLIPI published its first dedicated prognostic index — for advanced disease — in Blood.
This is the most current, prospectively-validated model in the field, derived from 552 patients with stage IIB–IVB disease.
It identified four important adverse factors at diagnosis:
N3 nodal status (effaced nodes)
Age >60
Raised LDH
Large-cell transformation in skin
| Risk group | Factors | 5-year OS |
|---|---|---|
| Low | 0–1 | 63% |
| Intermediate | 2 | 45% |
| High | 3–4 | 18% |
The headline message is that risk group, rather than stage alone, predicts survival in advanced disease
High-risk patients (3–4 adverse factors) should be considered for more aggressive treatment escalation — including allogeneic stem cell transplant assessment — regardless of their precise stage
Treatment of MF/SS
The aim of this section is to give a practical framework for thinking about treatment in MF/SS, rather than an exhaustive list of every agent. The British Association of Dermatologists (BAD) and UK Cutaneous Lymphoma Group guideline (Gilson et al., BJD 2019) and the BMJ Best Practice review (regularly updated) are good reference points if you want to dig deeper
A few principles guide everything that follows:
MF/SS is, in most cases, a chronic, incurable disease. The aim of treatment is to control disease, manage symptoms (especially pruritus), and preserve quality of life — not to achieve cure.
Stage drives treatment. Early-stage disease (IA, IB, IIA) is managed with skin-directed therapy. Advanced disease (IIB and above) usually requires systemic therapy, often combined with skin-directed approaches.
Aggressive systemic chemotherapy in early-stage disease does not improve survival and adds toxicity. The pivotal Kaye et al. study (NEJM 1989) randomised early-stage patients to combination chemoradiotherapy versus sequential topical therapy and showed no survival benefit — this finding still underpins current practice.
Allogeneic stem cell transplant is the only potentially curative treatment but carries significant treatment-related mortality and is reserved for younger, fitter patients with advanced disease.
Cutaneous lymphoma analogy for patients:
The immune system is a bit like a factory with workers spread out across the body. The factory is the bone marrow — that's where the immune cells are made. Once they're made, they get sent out to where they're needed: some go to the lungs, some go to the gut, and a particular group is sent to patrol the skin.
What's happened in your case is that a small group of those skin-patrol cells has started copying itself when it shouldn't. That's why the problem is showing up as patches on the skin — the cells that have gone wrong are the ones that live there. The factory itself, and the rest of your immune system, is working normally.
Because this is happening in the skin and not in the factory, it tends to stay a skin problem, often for many years. We don't usually have a cure for it, but the aim is to keep it under control — and you'll notice that many of the treatments we use are the same ones we use for eczema and psoriasis: creams, light therapy, sometimes tablets. That's not because we're underestimating it; it's because those are the treatments that work.
Our aim is to:
Improve symptoms, cosmetic appearances and quality of life
Reduce the incidence of recurrence
What we worry about is if the misbehaving cells start to leave the skin and travel back through the body — into the lymph nodes, or the blood, or further. That's why we keep a close eye on your nodes and your bloods at every visit.
Skin-directed therapy
Skin-directed therapy (SDT) is the mainstay of early-stage disease and remains an important adjunct at all stages, particularly for symptom control.
Skin-directed therapies
Topical agents
Phototherapy
Radiation therapy
Topical agents
Emollients and “expectant therapy”
In very early, asymptomatic disease, an "expectant policy" with emollients alone is reasonable — particularly in older patients, those with minimal cosmetic concern, and those where treatment side effects may outweigh benefit.
Reducing scale and dryness will reduce pruritus.
Topical corticosteroids
Usually first-line active treatment.
Potent or very potent preparations (e.g. clobetasol propionate) for ≈ 6 weeks.
Reported response rates in case series are high (around 80–90% partial response), but durability is poor and recurrence on cessation is common.
Maintenance use is often needed.
Zackheim et al. (Arch Dermatol 1998) reported a complete response in 63% of T1 patients and 25% of T2 patients at a median of 9 months' follow-up.
Topical tacrolimus 0.1%
Has limited supporting data in the literature but is clinically useful for face and flexural patch disease where steroid use is limited.
Topical mechlormethamine (nitrogen mustard)
A DNA alkylating agent which was historically compounded as an ointment
Now there is a 0.02% gel formulation (Valchlor / Ledaga) which is licensed by the FDA and EMA. Lessin et al. (JAMA Dermatol 2013) showed response rates of around 58% in stage IA–IIA disease
Side effects: burning, irritation, contact dermatitis (in around 30%); managed with concomitant topical steroids, dose reduction, or reduced frequency.
Small percentage in original studies developed NMSC but further long-term studies have not shown a clear increase in non-melanoma skin cancer with the gel formulation
Carmustine (BCNU) is an alterantive topical alkylating agent
Topical bexarotene 1% gel
FDA-approved but not licensed in Europe (as far as I am aware)
Overall response rate 63% and clinical complete response rate 21% in early stage CTCL (Brenean D et al. Arch Dermatol. 2002 March)
Side effects: Mild to moderate irritation at application site that often improves with use (like other topical retinoids)
Other topicals (limited evidence)
Topical imiquimod — small case series only
Phototherapy
Narrowband UVB (TL-01):
Effective for patch disease but limited dermal penetration means it is less effective for plaques.
One older study reported a complete response in 83% with median remission of 22 months (Ramsay et al., Arch Dermatol 1992).
The hypopigmented variant in younger patients tends to be particularly responsive
Relapse after stopping is common — sometimes within weeks — and patients should be counselled about this
PUVA:
Better penetration, so preferred for plaque-stage disease.
One of the most effective treatments for early disease, with response rates approaching 100% in some series.
More durable response than NBUVB, though formal head-to-head data are limited.
Can be combined with interferon-α or retinoids to improve response and reduce cumulative UV exposure.
Should cap at around 200 sessions lifetime to limit non-melanoma skin cancer risk
OTHER MODALITIES:
UVA-1 — small series, effective, but not widely available
Excimer laser (308 nm) — useful for localised disease
Clinical pearl:
Phototherapy is less effective in folliculotropic and syringotropic disease — the infiltrate sits too deep in the adnexa for UV light to reach
Radiotherapy
MF is one of the most radiosensitive tumours we treat. Radiotherapy can be used at every stage of disease
Localised superficial radiotherapy:
It can be used for isolated lesions and palliation of bulky plaques or tumours
Relatively low doses are needed, eg:
Patches and plaques: 8 Gy in 2 fractions
Tumours: 12 Gy in 3 fractions.
"Curative intent" doses (higher fractionation) can be considered for solitary lesions of pagetoid reticulosis or unilesional MF
More complex techniques (e.g. tomotherapy, HDR brachytherapy) are used for awkward anatomy — for instance if the lesion is wrapping around limbs, digits, or the face
Pearl — the abscopal effect: Occasionally, radiotherapy to one site is followed by improvement in untreated lesions elsewhere. This appears to be an immune-mediated phenomenon and is an active area of clinical trial interest
Total Skin Electron Beam Therapy (TSEBT)
Is a radiation treatment involving electrons that treats the entire skin surface
95% of the radiation only penetrates into the first 2cm of the body to spare vital organs from radiation
It can be used for widespread plaque or tumour disease refractory to other SDT
A low-dose regiment TSEB (12 Gy in 8 fractions over 2 weeks) is now generally preferred over a previous higher dose (Morris et al., IJROBP 2017):
Why low-dose is preferred:
Shorter, less toxic
Allows future re-treatment with TSEB if needed.
Can be combined with systemic agents.
Practical considerations: patients need to be able to stand for 30–40 minutes and lift their shoulders. Lead shielding for testes, eyelids, and other sensitive sites
Systemic therapy
Systemic therapy is generally indicated for:
Topicals and phototherapy are insufficiently effective
Relapse is rapid after phototherapy
Symptoms outweight the side-effect burden of escalation
Disease is widespread or located at particularly troublesome sites
Advanced-stage disease (IIB onwards).
A key counselling point: there is no robust evidence that systemic therapy in early-stage disease prevents progression or improves survival.
This is important to discuss with patients before escalation.
Choice of agent is guided by stage, comorbidities and patient preference
Systemic therapy
Established systemic agents
Newer targeted agents
Conventional chemotherapy
ESTABLISHED SYSTEMIC AGENTS
Bexarotene
Bexarotene (Targretin) is a synthetic rexinoid that selectively activates the retinoid X receptor (RXR) hence rexinoid.
Licensed indication: advanced MF refractory to at least one systemic therapy. But it is often used off-licence in refractory early-stage disease.
Efficacy
Phase II/III data: Early-stage disease: ORR 54% (n=58); Subsequent retrospective series report response rates up to 83%.
Response is dose-dependent, but dosing is limited by side effects.
Dosing
150-300mg/m2/day - typically 225-300mg/day starting dose and escalated 2-weekly as tolErated. Maximum response may take 6 months
Maintenance dose is typically 525-600mg/day
Adverse effects:
Hyperlipidaemia (≈ 83%) — triglycerides can climb to 10–12 mmol/L; Often may pretreat with fenofibrate prior to commencing it. May need to add other lipid lowering drugs if poorly controlled (eg statin, ezetimibe).
Central hypothyroidism (≈ 74%) — bexarotene suppresses TSH centrally. Pre-treat with levothyroxine (eg 25–50 mcg od) on day 1 of bexarotene. Monitor free T3/T4 and TSH together and adjust levothyroxine as required.
Neutropenia (≈ 47%).
Teratogenic — strict contraception required.
CYP3A4 interactions — including many lipid-lowering agents and azole antifungals.
Others - fatigue, dry skin/lips, headaches.
Baseline workup:
FBC, U&E, LFTs, lipid profile, CK, amylase, TFTs (TSH and T4), glucose.
Monitoring:
Two-weekly fasting bloods during dose escalations.
Interferon-α
Is a biological agent licensed for treatment of MF (can counsel patients this is not chemotherapy)
MoA is not not fully understood but it essentially wakes up the immune system and tips it back towards an anti-tumour response.
In MF, the malignant T cells are CD4+ helper cells with a Th2 profile — they produce cytokines that suppress the body's own anti-tumour immunity. Interferon-α counteracts this in three main ways:
Activates CD8+ cytotoxic T cells and NK cells — the cells that actually kill malignant lymphocytes
Suppresses the Th2 cytokine environment that the tumour cells thrive in, shifting the balance back towards a Th1 (anti-tumour) response
Increases visibility of the malignant cells to the immune system by upregulating MHC class I expression
Tend to use pegylated Interferon-α (subcut once weekly injection)
ORR ≈ 50% (up to 88%) in early-stage disease across multiple small non-randomised studies.
There are no formal MF/SS data for pegylated IFN, but efficacy is extrapolated from the non-pegylated formulations.
Adverse effects:
Flu-like symptoms after injection — pre-medicate with paracetamol 1 g 1 hour before
Depression — insidious onset over months; pre-existing depressive disorder is a contraindication
Thyroid dysfunction, hyperglycaemia
Neutropenia, transaminitis
Rare ocular changes — baseline eye test recommended
Caution with severe cardiac disease (HTN, arrhythmia, angina)
Methotrexate
Single-agent low-dose oral methotrexate has reported response rates around 76% with 5-year survival around 71% (older series).
It is a useful systemic option in MF/SS, particularly when an oral, lower-intensity treatment is preferred.
Higher response rates (based on retrospective studies) appear to be seen in erythrodermic MF rather than tumour-stage
Practical advantage: easier to monitor and prescribe than bexarotene or interferon
Extracorporeal photophoresis (ECP)
A "systemic form of PUVA".
Use in specialist centres.
It is primarily used in erythrodermic MF and Sézary syndrome, particularly where there is significant blood involvement.
It has a favourable tolerability profile and is often combined with interferon-α or bexarotene.
Responses may take several months, and it is generally less effective for bulky tumour-stage or transformed disease.
How it works:
Mononuclear cells are isolated by leukapheresis
They are then exposed to 8-methoxypsoralen and then UVA, and reinfused back into the patient
First-line for erythrodermic MF and Sézary syndrome
Can be used in combination with other treatments (eg IFN-α, bexarotene)
Typically given over 2 days every 2–4 weeks.
The UVA is directly toxic to irradiated cells and reinfused cells also appear to stimulate selective immune response against the malignant cells
Favourable factors for ECP response:
Short disease duration (preferably < 2 years)
Absence of bulky lymphadenopathy or major visceral involvement
White cell count < 20,000/μL
A discrete (not overwhelming) burden of Sézary cells (10–20% of mononuclear cells)
No prior intensive chemotherapy
NEWER TARGETED AGENTS
HDAC inhibitors (Vorinostat and Romidepsin)
Histone deacetylase (HDAC) inhibitors are systemic anti-cancer treatments, but they are better described as targeted/epigenetic therapies rather than conventional chemotherapy.
They work by altering how lymphoma cells switch genes on and off by inducing histone and protein hyperacetylation.
This can lead to cell cycle arrest and apoptosis.
Useful particularly for pruritus control as well as skin disease.
In the MAVORIC trial (see below), mogamulizumab was significantly superior to vorinostat for both PFS and ORR and vorinostat has been somewhat displaced as a standard comparator.
Brentuximab vedotin
Antibody–drug conjugate that targets CD30
ALCANZA trial (Prince et al. Lancet 2017): Phase 3 RCT, 131 patients with CD30+ MF (stage IB–IV) or pcALCL, randomised to brentuximab vs physician's choice (methotrexate or bexarotene):
Objective global response > 4 months: 56% vs 13%
Complete response: 16% vs 2%
Progression-free survival: 16.7 vs 3.5 months
Important practical points:
Access in Ireland and the UK requires documented CD30 positivity on biopsy.
CD30 expression in MF is variable across biopsy sites; multiple biopsies may be needed before CD30 positivity is obtained.
Initial response may be a flare; median time to response ≈ 12 weeks.
Useful as a bridge to alloHCT in advanced disease — small series show high response rates pre-transplant.
Adverse effects:
Peripheral neuropathy — reported in up to 45%, can be severe (motor and sensory). Dose reduce or stop early at the first signs.
Infusion reactions
Tumour lysis (reflecting how active the drug can be)
Drug rash, fatigue
Mogalizumab
A first-in-class defucosylated humanised anti-CCR4 monoclonal antibody.
CCR4 is highly expressed on malignant T cells in CTCL, particularly in blood-involved disease.
Defucosylation (removal of a sugar called fucose during manufacturing of the antibody) enhances antibody-dependent cellular cytotoxicity (ADCC) up to 100-fold compared with conventional mAbs.
MAVORIC trial — phase 3 RCT, mogamulizumab vs vorinostat in previously treated CTCL:
PFS: 7.7 vs 3.1 months (HR 0.53; p < 0.0001)
ORR: 28% vs 5% (p < 0.0001)
Significantly improved patient-reported quality of life (Skindex-29)
Particularly effective in Sézary syndrome and erythrodermic disease with blood involvement — response rates and durations are higher in blood/erythrodermic disease than in skin-only disease.
Adverse effects: Infusion reactions and drug eruptions (which can mimic disease progression — important to recognise).
Alemtuzumab
Anti-CD52 monoclonal antibody.
ORR ≈ 55% (32% CR) in advanced disease in older series. Reserved for refractory disease, generally after newer agents have failed.
Significant infection risk (CMV reactivation, opportunistic infection) — careful monitoring required.
Conventional chemotherapy
Chemotherapy is generally reserved for advanced, rapidly progressive, transformed, or refractory MF/SS after better-tolerated systemic options have been considered.
Combination chemotherapy, such as CHOP or EPOCH, can produce high initial response rates, but responses are usually short-lived, toxicity is significant, and there is no clear survival benefit.
When chemotherapy is needed, single-agent treatment is usually preferred over combination regimens. Options include gemcitabine, doxorubicin and purine analogues such as pentostatin, cladribine or fludarabine.
In general, chemotherapy in MF/SS is palliative rather than curative.
CD30+ve Lymphoproliferative disorders
CD30 is a protein made by activated T and B lymphocytes
It is present in CD30 +ve lymphoproliferative disorders, some cases of MF and Hodgkin’s lymphoma
After mycosis fungoides, the CD30+ lymphoproliferative disorders are the second commonest group of CTCLs (~25%). They sit on a spectrum and share CD30 expression, but differ markedly in clinical behaviour. The two main entities are:
Lymphomatoid papulosis (LyP) — chronic, self-healing papulonodular eruption with malignant histology but benign course
Primary cutaneous anaplastic large cell lymphoma (pc-ALCL) — solitary or localised tumour(s) with an excellent prognosis
Both must be distinguished from transformed MF (where CD30+ large cells appear within an established MF backdrop) and from systemic ALCL with secondary skin involvement — clinically essential because management and prognosis differ substantially.
Both must be distinguished from transformed MF (where CD30+ large cells appear within an established MF backdrop) and from systemic ALCL with secondary skin involvement — clinically essential because management and prognosis differ substantially.
Key point — CD30 positivity is not specific. Reactive infiltrates (arthropod bites, scabies, drug reactions, PLEVA, herpes/orf) can contain CD30+ cells. Diagnosis requires clinicopathological correlation, not histology alone.
Lymphomatoid papulosis
Commonest ‘CTCL’ in children but age range is wide (average age of onset 35-45)
Clinical features:
Chronic, recurrent crops of papules and small nodules, typically <2 cm
Lesions characteristically crust, ulcerate and self-heal over 3–12 weeks, often leaving varioliform/atrophic scars
Trunk and limbs are most commonly affected; but can occur anywhere including the palms and soles
Lesions at different evolutionary stages coexist
Asymptomatic or mildly pruritic
Clinical ddx - insect bites, acne, PLEVA, molluscum
Histopathological subtypes (WHO 5th edition)
LyP can be histologically diverse but generally all variants have an excellent prognosis
Lymphomatoid papulosis — histological subtypes
A folliculotropic variant ("type F") has been described but is not universally accepted.
Pearl — No subtype carries a worse prognosis. Subtyping matters for histological recognition, not for clinical decision-making.
Immunophenotype & clonality
CD30+ large atypical cells (membranous and paranuclear/Golgi dot-like staining)
Usually CD4+, CD8−; pan-T cell markers often partially lost
ALK negative, EMA negative(distinguishes from systemic ALCL)
A T-cell clone is found in ~60–70% — although clonality is not required for diagnosis, and absence does not exclude LyP
Diagnosis
LyP is fundamentally a clinicopathological diagnosis: the clinical course of relapsing, spontaneously regressing lesions is essential. Histology alone cannot make the diagnosis — type C, in particular, is indistinguishable from pc-ALCL on biopsy.
Pearl — If you can't see the patient's lesions resolving spontaneously, you can't confidently diagnose LyP.
Prognosis
Excellent — close to 100% 5-year disease-specific survival
However, approximately 10–20% develop an associated lymphoma at some stage in their life, including:
Most commonly MF (often sharing the same clone)
pc-ALCL, less commonly
Less commonly - Hodgkin disease or systemic ALCL (
The associated lymphoma may develop decades after LyP onset — lifelong follow-up is recommended
Management
The mainstay is watchful waiting: most patients need no active treatment beyond reassurance and education.
When intervention is warranted (cosmetic burden, frequent recurrences, scarring):
Topical corticosteroids — short-lived response
Phototherapy (NB-UVB or PUVA) — reduces lesion frequency; relapse common on discontinuation
Low-dose methotrexate (5–25 mg weekly) — most effective long-term option; well tolerated
Brentuximab vedotin — reserved for refractory/severely symptomatic disease (off-label in LyP, but shown to be active in phase 2 studies)
Avoid systemic chemotherapy — it is not indicated and does not alter prognosis
Primary cutaneous anaplastic large cell lymphoma (PC-ALCL)
Part of the spectrum of CD30+ve CTCL
Typically get a solitary tumour which may increase in size and ulcerate
Histopathology:
Histological features of PC-ALCL can resemble those seen in anaggressive systemic lymphoma but the disease doesn’t follow an aggressive course
Confluent sheets of large, atypical lymphocytes infiltrating dermis and subcutis
Don’t tend to see epidermotropism
Mitotic figures abundant
Other ddx:
Can share histological features with Lymphomatoid papulosis
MF can show large cell transformation and can be CD30+ve (if have multiple patches and plaques consider MF)
Immunohistochemistry:
CD30+ve cellswhich can be CD4+ or CD8+ve
(may also express CD3 and CD5)
It is essential to distinguish primary cuataneous anaplastic large cell lymphoma (PC-ALCL) from systemic ALCL
The absence of staining with anaplastic lymphoma kinase (ALK) favours the primary cutaneous variant
(Think the absence of staining means it is absent elsewhere)
ALK staining is detected in > 90% of systemic ALCLs and results from a translocation involving the ALK gene causing the expression of the ALK protein
(Translocation is detectable with cytogenetic tests)
Treatment:
Lesions can be excised or treated with radiotherapy
Spontaneous regression may occur
Multiple cutaneous lesions or nodal spread may require systemic treatment
Brentuximab vedoin (Adcetris):
Granted FDA approval November 2017 for treatment of adult patients with:
Primary cutaneous anaplastic large cell lymphoma or CD30 +MF who have received prior systemic therapy
ALCANZA trial:
Phase 3 trial
Looked at patients with CD 30+ cutaneous T-cell lymphomas (either CD30+ MF or pc-ALCL)
A significant improvmeent in objective global response was seen with brentuximab versus physician’s choice of mehtotrexate or bexarotene
Objective global resposne: 56% vs 13%
Complete response: 16% vs 2%
Progression free survival: 16.7 months vs 3.5 months
B Cell Lymphoma
Primary cutaneous B cell lymphomas account for ≈ 20-25% of primary cutaneous lymphomas
The clinical picture is different from CTCL. You don't get the scaly patches and eczema-like plaques of mycosis fungoides.
Instead, B-cell lymphomas present with plum-coloured or red-brown, dome-shaped nodules or plaques
In most cases they are asymptomatic
Can be isolated lesions or a few scattered lesions
They can be divided up as follows:
Site is a useful first clue:
Indolent CBCLs (follicle centre, marginal zone) favour the head/neck and trunk.
Whereas the aggressive entity DLBCL, leg type favours the lower legs.
Immunohistochemistry - keeping it simple
The principle is identical to the T-cell side
Cells carry CD markers (proteins) on their surface and immunohistochemical stains are antibodies that latch onto a chosen marker and stain it brown, so we can see which cells are present and whether they're behaving normally.
For B cells, the master flag is CD20.
Once CD20 confirms you're dealing with a B-cell infiltrate, a small panel sub-types it
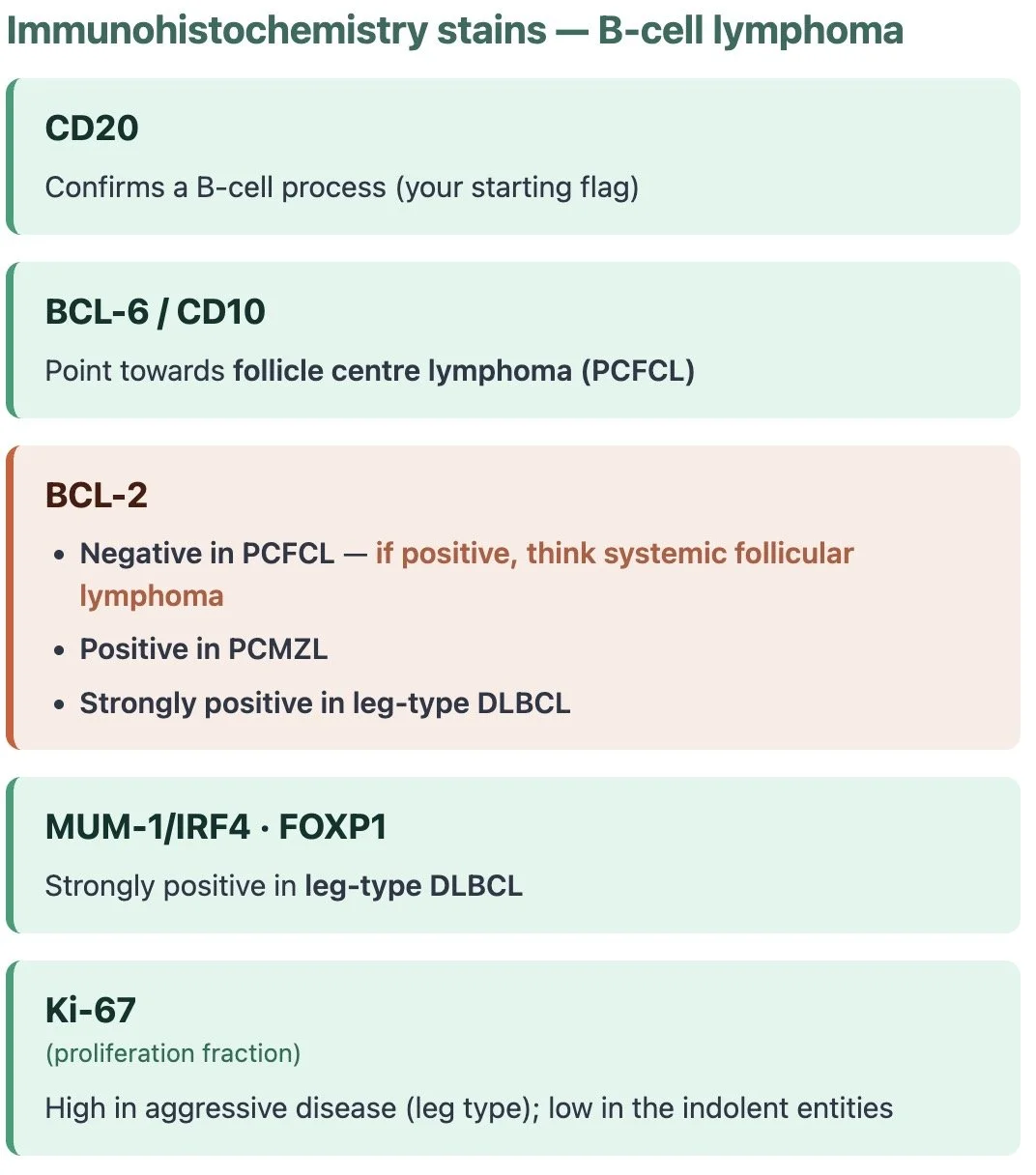
The immunohistochemistry of PCFCL tends to be a mirror image of PCMZL

Clinical pearl:
One to be aware of is BCL-2 — it is usually negative in cutaneous follicle centre lymphoma but positive in systemic follicular lymphoma so if it looks like a follicular cell lymphoma but is BCL-2 positive you need to really think about systemic follicular lymphoma
Clonality:
Every B cell carries its own unique antibody (immunoglobulin). A normal reactive infiltrate is therefore polyclonal — a crowd of B cells, each with a different antibody. In lymphoma, a single clone has expanded and taken over, so that diversity collapses. Demonstrating that collapse is what clonality testing is all about.
Just as T cells have a T-cell receptor, B cells carry a B-cell receptor (BCR) — essentially the membrane-bound version of the immunoglobulin the B cell would otherwise secrete. We interrogate it two complementary ways.
1. Immunoglobulin gene rearrangement (IGH + IGK)
This is the B-cell equivalent of TCR gene rearrangement. PCR is used to amplify the rearranged immunoglobulin heavy chain (IGH) and kappa light chain (IGK) genes; a single dominant (monoclonal) peak standing out against the normal polyclonal background suggests a B-cell lymphoproliferative process.
Test both genes. Testing IGH and IGK together pushes the detection rate close to 99%, and IGH alone can give false negatives.
2. Light chain restriction
A simpler, complementary test, usually done by immunohistochemistry or flow cytometry. Every B cell makes antibody with either a kappa or a lambda light chain — never both. In a normal reactive infiltrate you see a mixture of kappa- and lambda-expressing B cells in a roughly 2:1 ratio.
If virtually all the B cells express the same light chain — the ratio is heavily skewed, i.e. "kappa-restricted" or "lambda-restricted" — that tells you they are all descended from a single parent cell, and supports a clonal B-cell process.
Follicle centre vs marginal zone: a quick refresher
B cells live and mature in lymphoid tissue — eg lymph nodes, spleen and gut
Lymph node
Within lymphoid tissue you find follicles, and each follicle has different zones.
A naïve B cell waits in the follicle until it meets its antigen. When it does, it moves into the germinal centre to be refined — maturing through centroblasts (large, rapidly dividing cells) into centrocytes (smaller cells).
Having matured, the experienced cell then moves out to the marginal zone (the outer ring) as a memory or plasma cell.
This matters because each lymphoma mirrors the cell it's stuck at:
BCL-6 and CD10 are germinal-centre markers — if positive, they point to a follicle centre lymphoma (PCFCL).
Marginal zone lymphoma (PCMZL) is a post-germinal-centre cell, so it tends to be BCL-6− and CD10−, and you may also see clones of plasma cells.
Primary cutaneous follicular cell lymphoma
The commonest CBCL — roughly 55% of cases.
Firm pink-to-plum grouped papules, nodules and plaques on the head, neck and trunk (classically scalp/forehead and back).
Scalp lesions may be associated with alopecia
Slow-growing and indolent;
Multiple lesions and local relapse are common but systemic spread is rare
Histology: proliferation of centrocytes (± centroblasts) in a follicular, mixed or diffuse pattern in the dermis, sparing the epidermis (no epidermotropism).
IHC:
CD20+
BCL-6+
CD10 often +
Crucially BCL-2 negative (or only weakly positive). This is where your BCL-2 pearl pays off — a follicle-centre-looking lymphoma that is strongly BCL-2+ should make you think systemic follicular lymphoma with skin spread, so stage them.
Prognosis: excellent, 5-year survival >95%.
Treatment: local radiotherapy or excision for localised disease; rituximab or watchful waiting for multifocal disease.
Always stage to exclude secondary cutaneous involvement by nodal follicular lymphoma.
Primary Cutaneous Marginal Zone Lymphoma
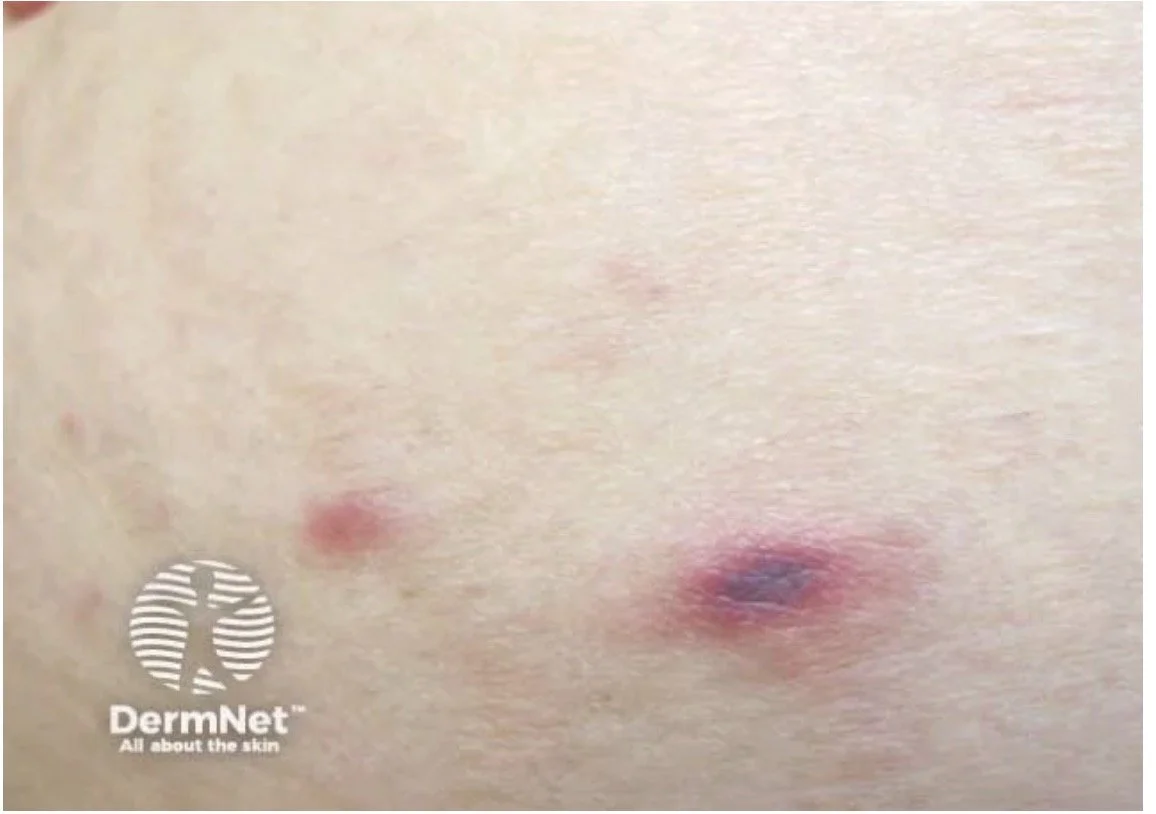
Cutaneous MALT lymphoma. PCMZL is essentially the skin's version of a MALT lymphoma — a low-grade, marginal-zone B-cell lymphoma arising in extranodal tissue rather than a lymph node. As a reminder, mucosa-associated lymphoid tissue (MALT) is the immune tissue lining organs exposed to the outside world (e.g. gut, lung). Unlike those sites, the skin has no organised MALT — here the lymphoid tissue is acquired, sometimes driven by a chronic trigger (e.g. Borrelia in endemic areas, arthropod bites).
Clinical:
The youngest-presenting cutaneous B-cell lymphoma, typically in younger to middle-aged adults.
Solitary or, more often, multifocal red-to-violaceous papules and nodules, favouring the trunk and upper limbs.
Usually asymptomatic; lesions may wax and wane or partially regress and rarely ulcerate.
Local skin relapse is common, but extracutaneous spread is rare and behaviour stays indolent.
Borrelia burgdorferi has been implicated in endemic European areas only (geographically variable and debated).
Histopathology & immunohistochemistry:
A nodular or diffuse dermal infiltrate of small marginal-zone (centrocyte-like) cells, lymphoplasmacytoid cells and peripheral plasma cells, around reactive germinal centres.
The profile is the mirror image of PCFCL: CD20+, BCL-2+, BCL-6−, CD10−. Clonality is usually confirmed by light-chain (kappa/lambda) restriction in the plasma-cell component
Treatment:
Observation
Intralesional steroids
Local radiotherapy
Excision
Rituximab if extensive
A doxycycline trial is reasonable where Borrelia serology or PCR is positive.
Prognosis:
Excellent, with 5-year survival well over 90%.
Extracutaneous spread is rare and large-cell transformation exceptional.
Pearl —
There's genuine overlap with cutaneous lymphoid hyperplasia (pseudolymphoma), which is why it is increasingly reframed as a lymphoproliferative disorder — the ICC 2022 labels it primary cutaneous marginal zone lymphoproliferative disorder rather than a frank lymphoma.
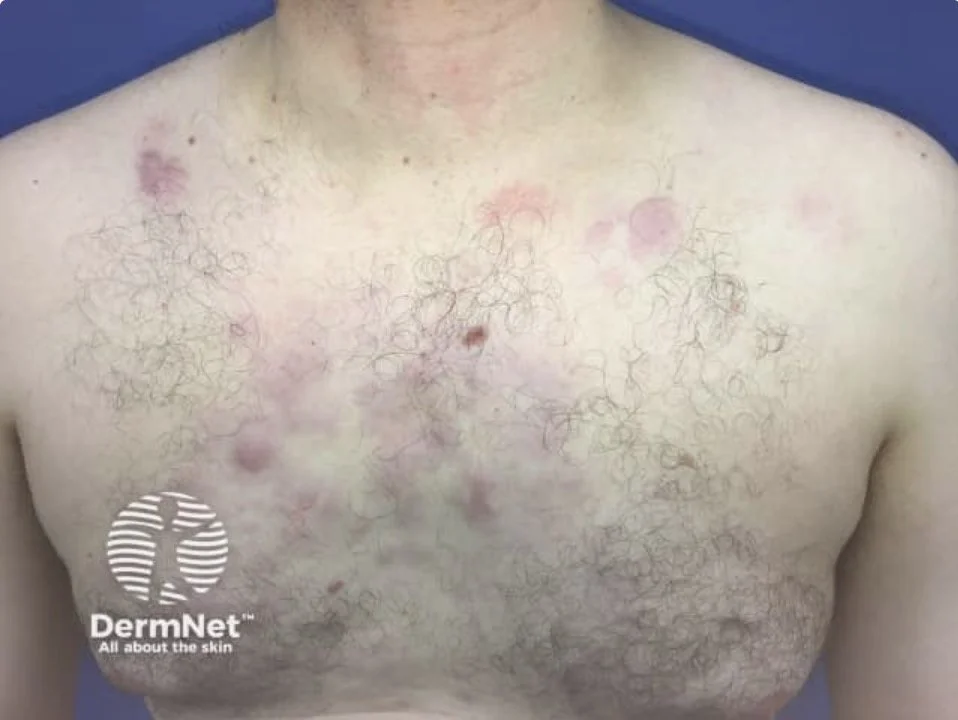
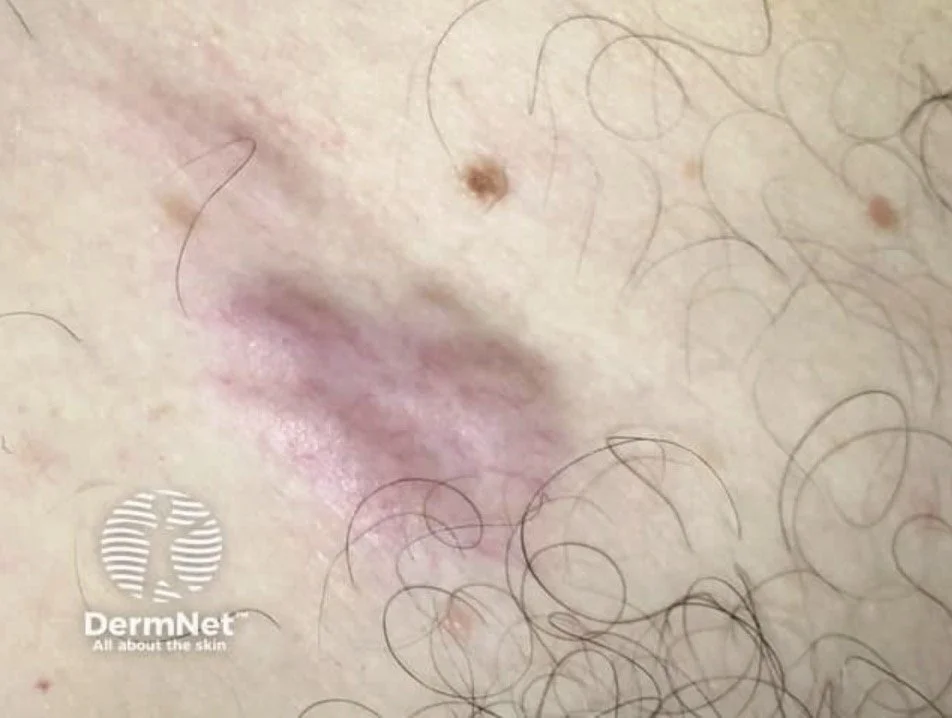
Primary Cutaneous diffuse large B cell lymphoma - leg type
This is the aggressive primary cutaneous B cell lymphoma that we should be aware of
Clinical.
Elderly patients (median age ~76), with a female predominance. Rapidly growing red-brown or bluish nodules and tumours, usually on one or both lower legs; about 10–15% arise elsewhere.
Histopathology & immunohistochemistry.
Diffuse dermal sheets of large centroblasts and immunoblasts with a high mitotic rate. The immunophenotype is CD20+, strongly BCL-2+, MUM-1/IRF4+, FOXP1+, CD10−, with variable BCL-6 and a high Ki-67 — effectively the cutaneous counterpart of activated B-cell (non-germinal-centre) DLBCL. The MYD88 L265P mutation is present in up to around 70% of cases and marks a poorer outcome.
Treatment.
Treated like systemic DLBCL: R-CHOP ± involved-field radiotherapy. Frail or elderly patients unfit for chemotherapy are considered for palliative radiotherapy. Urgent haematology referral.
Prognosis.
5-year disease-specific survival ~50–60% (improving towards 60% with R-CHOP).
Pearl — This is the cutaneous B-cell lymphoma that behaves like a systemic one, so it's staged and treated as one. Site plus age is the tell: a rapidly growing nodule on the lower leg of an elderly woman is leg-type until proven otherwise.
BLASTIC PLASMACYTOID DENDRITIC CELL NEOPLASM (BPDCN)
A rare, clinically aggressive neoplasm — not a B- or T-cell lymphoma.
BPDCN is derived from the immature precursors of plasmacytoid dendritic cells (pDCs), so despite sitting in the cutaneous lymphoma world it isn't a lymphocyte tumour at all.
pDCs are a rare type of immune cell (potent producers of type I interferon) that arise from bone-marrow haematopoietic stem cells.
BPDCN was previously called blastic NK-cell lymphoma and CD4+/CD56+ haematodermic neoplasm
Epidemiology:
Male predominance (M:F ≈ 3.3:1), usually in the 7th–8th decade, though it can occur at any age.
There is a recognised association with myelodysplastic syndromes and myeloproliferative neoplasms (particularly chronic myelomonocytic leukaemia).
Clinical:
BPDCN tends to involve multiple sites — skin (60–100%), bone marrow and peripheral blood (60–90%), and lymph nodes (40–50%).
The skin is usually the first site affected (~90%), and disease often remains confined there before a rapid second phase of leukaemic spread and multi-organ involvement — the skin behaving almost as a "sanctuary" site that limits early dissemination.
Lesions are bruise-like: erythematous-to-purplish papules, plaques or tumours of varying size, with no preferred anatomical site; they may be solitary or widespread, and mucosal involvement is rare. Isolated lesions carry a better progression-free survival (~23 months) than eruptive, disseminated disease (~9 months).
Histopathology & immunohistochemistry:
A diffuse, monomorphous dermal infiltrate of medium-sized cells with immature chromatin, resembling lymphoblasts or myeloblasts. The dermis is typically massively infiltrated with extension into subcutaneous fat, while the epidermis and adnexa are spared.
The diagnostic immunophenotype is the classic triad — CD123, CD4, CD56 — usually with TCL1 positivity.
Crucially, lineage markers are negative: CD19/CD20 (B-cell), CD3 (T-cell) and myeloperoxidase (myeloid) — which is what excludes the mimics.
Immunohistochemistry — BPDCN
Positive
the diagnostic triad, plus TCL1
Negative
excludes the mimics
Treatment:
Historically chemoresistant, with only transient responses and a median overall survival of roughly one year.
Management is haematology-led and has changed in recent years:
Tagraxofusp (a CD123-directed agent) is now the approved first-line therapy — BPDCN uniformly expresses CD123.
Intensive ALL- or AML-like induction regimens are alternatives.
Allogeneic stem cell transplant in first remission offers the only reliably durable survival; tagraxofusp used to bridge fit patients to transplant is now the preferred pathway.
Urgent haematology referral.
Prognosis:
Poor overall, though improving.
Median overall survival was historically ~1 year, and only patients who reach allogeneic transplant achieve prolonged survival (some series report 3–4-year overall survival of 75–80% after transplant).
Topics to do:
Subcutaneous panniculitis like T cell lymphoma:
Commonest cause of panniculitis is E nodosum
What are the things that make panniculitis atypical:
Persistance vs recurrent episodes with complete clearing in between
Tend to be on lower extremities also
On pathology tends to be a lobular panniculitis (similar to lupus panniculitis)
Will have atypical cells on H&E
Can be difficult to distinguish between lupus panniculitis and subcutaneous panniculitis like T cell lymphoma
Some consider them to be two entities in a spectrum
About 20% of patients with subcutaneous panniculitis T cell lymphoma have positive ANA
Staging cutaneous lymphomas other than MF/SS:
In 2007, a staging system was developed for all ohter cuatnous lymphomas that describes the extent of the lymphoma (using a TNM classification) but it does not give any useful information about prognosis
I won’t go into much more detail about that here
Issues:
Many different type of cutaneous lymphomas
Most are rare
Not useful in clinical practice
Reason for classification:
Current stating system helps classify patients
Over time may be able to better define prognosis for different types of cutnaeous lymphoma
Currently it is easier to define prognosis based on growth pattern of the lymphom
Indolent:
Grows slowly
Treatment may not be necessary unless causing symptoms
Often not curable

Aggressive:
Grow rapidly
Treatment necessary for survival
Often curable

Parapsoriasis:
Pseudolymphoma:
Term represents several clinical entitiies that probably have multiple aetiologies
Lymphocytoma
Spiegler-Fendt sarcoid
Lymphadnosis benigna cutis
Jessner’s benign lymphocytic infiltrate
In most cases aetiology is unknown (some cases may be related to chronic arthropod bites)
Usually present as indloent single or grouped red/purple nodules or plaques on the head, neck and upper trunk
Infiltrate may show B or T cell predominance
Some cases can be difficult to differentiate from lymphoma and patient should be follwed up prior to definitive diangosis being made
Leukaemia cutis:
CLL in adults is the most common cause of specific leukemic skin lesions in adults
They are usually multiple and may present wih papules, nodules, plaques, erythema and rarely bullae
The neoplastic proliferation of lymphocytes is usually B cell in origin