
Clinical appearance (based on size of vessel affected)
Small vessel vasculitis:
Cutaneous small vessel vasculitis
Small and medium vessel vasculitis:
Granulomatosis with polyangiitis (Wegener’s)
Eosinophilic granultomatosis with polyangiitis (Churg Strauss)
Medium vessel vasculitis:
INTRODUCTION
Vasculitis is inflammation directed towards the blood vessel wall
It can occur in any organ of the body
It can be limited to the skin or it can be a manifestation of a systemic vasculitis
Can get vasculitis of arteries, veins, capillaries
How it presents depends on the size of blood vessel affected and current classification is based on the size of the blood vessel involved
Cutaneous involvement in the majority of cases tends to affect the small and medium sized vessels
PATHOGENESIS
Terms:
Cutaneous small vessel vasculitis (CSVV) refers to a vasculitis involving primarily dermal post-capillary venules and when a biopsy is taken you see a specific histological pattern of vasculitis called leucocytoclastic vasculitis (LCV)
In LCV the postcapillary venules of the dermis are affected by an intense neutrophilic vascular inflammation
LCV often used interchangeably with CSSV but LCV however:
LCV can be seen in settings of mixed small and medicum vessel vasculidities (eg Granulmatosis with polyangiitis) whereas the term CSVV should generally be reserved for when only small vessels are involved (without any mixed sized vessel involvement)
Leucocytoclastic vasculitis on biopsy:
Neutrophils around blood vessels
Leucocytoclasis (degnerating neutrophils) forming ‘nuclea dust’
Fibrinoid necrosis (within vessel wall, looks pink)
Extravasated red cells
Small and medium vessel vasculidities tend to be either an immune complex mediated vasculitis or an ANCA associated vasculitis
Immune complex mediated vasculitis pathology:
All forms of CSVV and some forms of mixed and medium-sized vessel vasculidities are mediated by immune complexes (type 3 hypersensitivity reaction)
Essentially an antigen (eg a virus) is bound by antibodies and forms immune complexes
These deposit into postcapillary venules in the skin
These lodged complexes then activate complement which attracts neutrophils which damage the blood vessels allowing red blood cells to leak out
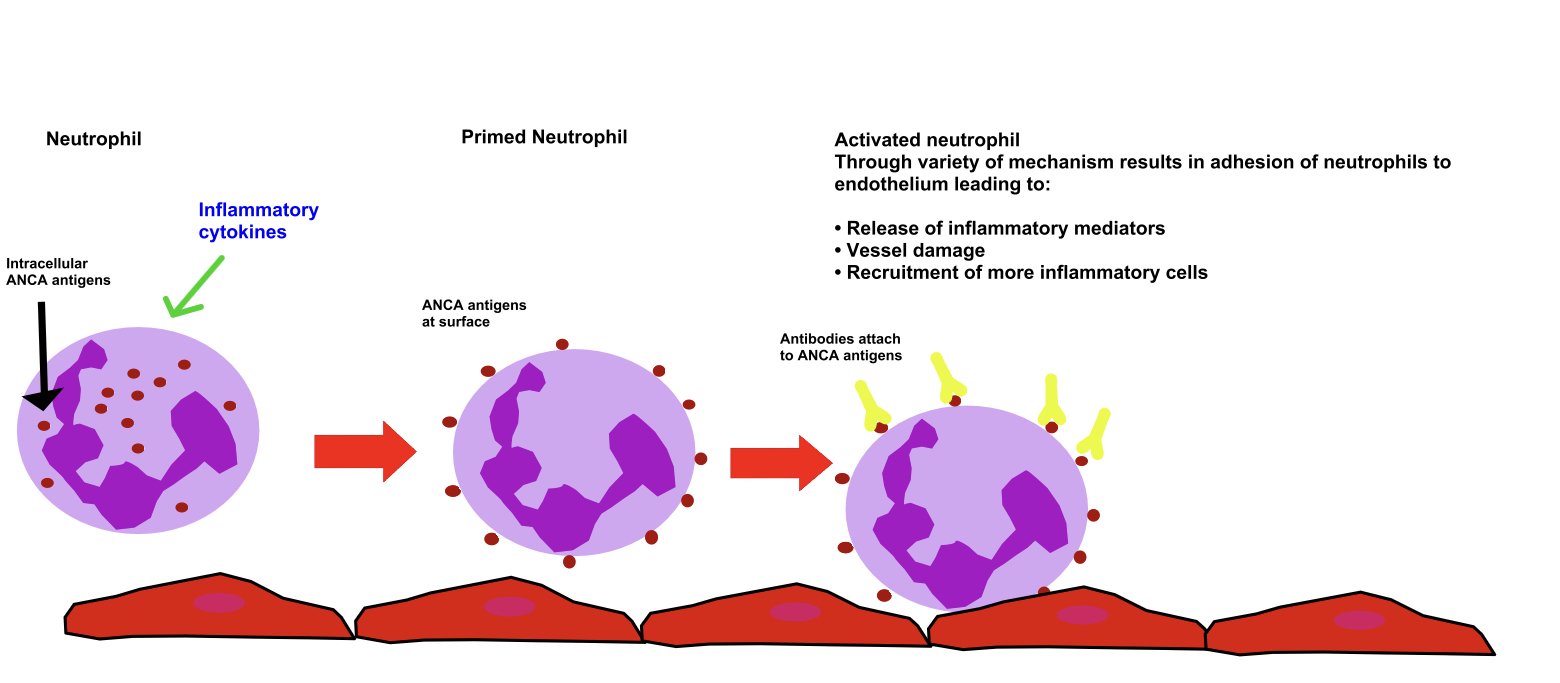
ANCA associated vasculitis pathology:
The ANCA associated vasculidities are not mediated by immune complexes
PR3 and MPO are intrecellular neutrophillic proteins that can translocate to the neutrophil cell surface after activation by cytokines (eg TNFα)
An ANCA antibody can then bind to these antigens resulting in adhesion of neutrophils to the blood vessel endothelium
This results in release of inflammatory mediators, vessel damage and recruitment of additional inflammatory cells
As there is a lack of immune complex deposition on immunoflourescence we call this pauci-immune

CLINICAL APPEARANCE (based on size of vessel affected)
Small Vessels:
Arterioles, capillaires, postcapillary venules
Superficial and mid dermis
Skin manifestations:
Palpable purpura
If gently feel around area, it will feel raised
Non-blanching
Macular purpura
Petechiae
Urticarial papules
Vesicles
Pustules
Targetoid papules and plaques

Dr. McColl

Palpable purpura
Courtesy Dr. Mccoll
Medium Vessels:
Small arteries and veins
Deep dermis and subcutis
Skin manifestations:
Livedo racemosa
Retiform purpura
Ulcer
Subcutaneous nodules
Digital necrosis
So can get a patient with manifestations of:
Small vessel signs only
Medium vessel signs only
Combination of both
CLASSIFICATION
Clinically (+/- histologically) should give consideration to what size of vessels are affected as this may give a clue to the underlying cause
Classification by size (Chapel Hill classification)

Small vessels:
Idiopathic cutaneous small vessel vasculitis
Secondary cause of cutaneous small vessel vasculitis
Drugs
Infection
Malignancy (usuallly haematologic)
Urticarial vasculitis
Henoch-schonlein purpura
Acute haemorrhagic oedema of infancy
Erythema elevatum diutinum
Small and medium vessels:
Cryogloulinaemia (Type II and III)
ANCA-associated
Microscopic polyangiitis (MPA)
Wegener’s granulomatosis (GPA)
Churg-Strauss (EGPA)
Secondary causes
Infection
Inflammatory disorder (eg autoimmune connective tissue disease)
Medium sized vessels:
Poyarteritis nodosa (PAN)
Classic (systemic) PAN
Cutaneous PAN
Large sized vessels:
Temporal arteritis
Takayasu’s arteritis
Other considerations when classifying cutaneous vasculitis:
Look for underlying causes
? ANCA positive or negative
Direct immunoflouroescence findings: Especially ? IgA predominance
Evidence of systemic manifestations
REVIEW OF SYSTEMS
If there is concerns about a systemic vasculitis a thorough review of systems should be performed in history for past and present symptoms.
Constitutional: Fever, weight loss, night sweats, arthralgias
URTI
Oral/nasal ulcers
Nasal perforation, saddle nose deformity
Sinusitis
Hearing loss/recurrent otitis media
Change in voice (vocal cords)
Subglottic stenosis
Eyes
Psueotumours
Conjunctivitis
LRTI (lung infiltrates, cavities, haemorrhage)
Can be sudden and acute or chronic
Shortness of breath, cough haemoptysis, pleuritis
Cardiac (myocarditis/pericarditis)
Chest pain, shortness of breath, palpitations
GIT:
Abdominal pain
Change in bowel habits
Blood pr
Kidneys: (Glomerulonephritis, vascular occlusion)
Urine dipstick blood, protein, casts
Urine Protein:creatinine ratio
Peripheral neuropathy
Motor (eg foot drop)
Sensory
CNS changes
Stroke like symptoms
Skin changes
Palpable purpura
Livedo reticularis
Nodules
Ulceration
Necrosis
VASCULITIC SCREEN
When assessing for a vasculitis it is important to look for any kidney damage and is essential to check and monitor blood pressure and perform a a urine dipstick
If the urine dipstick shows protein and/or blood it is important to send for urinary protein:creatinine ratio
Also may perform ‘vasculitic screen’, an example of a vasculitic screen that could be considered would be:
FBC
U&E/LFT
CRP/ESR
Antibodies: ANA, ENA, dsDNA, RF
Anti-neutrophil cytoplasm antibodies (ANCA)
Complement levels
Cryoglobulins
Hepatitis B/C, HIV serology
Immunoglobulins
Serum electrophoresis
Anti-streptocccal titre (ASOT)
Skin biopsy with samples sent for H&E and direct immunofluorescence (DIF)
Ideally performed in first 24-48 hours
DIF:
+ve in 80% CSVV
+ve 100% lesions < 24 hours old
+ve 30% at 48-72 hours
Only C3 > 72 hours
Granular pattern within vessel wall
C3, IGM, IgA +/- IgG
Negative in ANCA associated vasculitis
CUTANEOUS SMALL VESSEL VASCULITIS (CSVV)
As mentioned previously it involves primarily dermal post-capillary venules and it is charachterised histologically by a leucocytocastic vasculitis
The term CSVV is generally reserved only for when small vessels are involved (without medium sized vessel involvement )
Causes:
Idiopathic (45-55%)
Infection (15-20%)
Bacterial - Group A strep
Viral - Hepatitis B/C, HIV
Fungal - Candida
Inflammatory (15-20%)
Autoimmune onnective tissue disease: RA, SLE, Sjogen’s
IBD
Seronegative spoindylarthropathy
Drug (10-15%) (following list not exhaustive)
Anti-inflammatories:
NSAIDs
COX-2 inhbitors
Antibiotics:
Beta lactam antibiotics
Trimethoprim/sulfamethoxazole
Quinolones
Vancomycin
Allopurinol
Cardiac medications
Diuretics: thiazides, loop
ACEi, beta blockers
OCP
Neoplasm (5%)
Often haematological but can be due to solid organ cancers also
Within the spectrum of CSVV there are several subtypes with unique features:
Henoch schonlein pruprua
Acute haemorrhagic oedema of infancy
Urticarial vasculitis
Erythema elevatum diutinum
CSVV clinical features:
Typically present 1-2 weeks after exposure to inciting agent
Usually presents with palpable purpura and petechiae on dependant areas such as the lower legs and pressure points
Can also present as:
Urticarial lesion
Vesicles
Pustules
Targetoid lesions
Lesions often more severe in areas under pressure (eg sock line)
Can be itchy +/- painful
Residual postinlfammatory hyperpigmentation may persist for months after primary process resolves
Extracutaneous involvement can occur (eg fever/arthralgias, gastrointestinal or genitourinary involvement) but is uncommon and usually mild
Prognosis:
90% will have spontaneous resolution of cutaneous lesions within weeks/few moths
10% will have chronic or recurrent disease at intervals of months to years
Assessment:
Determine if patient has particular sub-type of CSVV (eg HSP)
Outrule a systemic vasculitis syndrome
Consider secondary causes
Treatment:
It often resolves without any treatment
Remove inciting trigger
Supportive measures:
Leg elevation, avoid tight clothing, rest
Symptomatic therapy:
Antihistamines, NSAIDs
Chronic disease (> 4 weeks) or more severe cutaneous disease consider:
Colchicine 0.6mg bd-tds (often get GI side effects)
Dapsone 50-200mg/day
Can consider both in combination
Severe, ulcerating or progressive cutaneous disease needing rapid control:
Short course of systemic oral steroids
Up to 1mg/kg/day
Taper over 4-6 weeks
If recurs when dose is tapered or disease is recalcitrant may need steroid sparing agent
Methotrexate
Azathioprine
HENOCH SCHONLEIN PURPURA
HSP is an IgA vasculitis (IgA predominance seen on immunoflourescence)
It is the commonest form of vasculitis in children
(90% occur in children < 10 years)
Children classically initially have a URTI or strep infection
1-2 weeks later symptoms and signs
Evidence of medium vessel disease or widespread lesions (including face) could indicate an underlying IgA paraproteinaemia
HSP tetrad:
1. Palpable purpura legs and buttock
2. Arthralgia of knees and ankles
3. GI issues including abdominal pain and diarrhoea with or without melena
4. Renal changes with haematuria, possible nephritis and rarely renal failure (1% of cases)
HSP clinical findings:
Cutaneous findings:
Erythematous papues/plaques evolving to palpable purpura is classical finding
Urticaria, vesicles, bullae and foci of necrosis can be seen
Typically symmetrical and distributed on buttocks and lower extremities
Can involve trunk, upper extremities, face
Individual lesions last 10-14 days with skin resolution over weeks-months
Can get recurrent skin disease in 5-10% of patients
Arthritis:
Up to 75% of patients
Commonly joints of lower extremities (knees and ankles)
Gastrointestinal::
50-75% of patients
May precede purpura
Colicky abdominal pain
Vomiting
GI bleeding (30%)
Rarely - intussusception and bowel perforation
Renal:
40-50% of patients
Microscopic haematuria (40$)
Proteinuria (25%)
Cutaneous lesions often precede nephritis but latter usually clinically evident within 3 months
Persistent renal disease (requiring monitoring till resolution) in 30-50% of patients
Only 1-3% of children develop long-term renal impairment
Orchitis:
Can rarely occur in young boys
Poor prognostic features HSP:
Renal failure at time of onset
Nephrotic syndrome
Hypertension
IgA small vessel vasculitis adults (sometimes called adult HSP)
Clinical presentation and prognosis differ to children
Necrotic skin lesions present in 60% of adults (vs <5% children)
Up to 30% may develop chronic renal insufficiency
3 findings suggestive that an adult with HSP will have kidney involvement with a IgA glomerulonephritis:
1. Fevers
2. Increased ESR
3. Purpura located above the waist (‘think purpura getting close to kidneys so more likely to be involved’)
Adults more likely to require more aggressive therapy
If IgA vasculitis due to cancer 60-90% is due to solid organ cancer (particularly lung)
This is in contrast to most CSSV in which cancer is usually haematologic
Diagnosis:
Biopsy: leucocytoclastic vasculitis
DIF: IgA deposition in blood vessel walls so many people recommend to do biopsies in rashes suspicious for vasculitis as it will give an indication if we need to monitor for renal disease as time goes by
(IgA can be seen in other types of CSSV so diagnosis of HSP supported by IgA predominance)
Monitoring:
Serial urinalysis (+/- urine pcr)
Check faecal occult blood if have GI symptoms
Treatment:
Disease usually self-limited and resolves over weeks to months
Supportive
Dapsone or colchicine may decrease duration of cutaneous lesions
Systemic steroids can treat the arthritis, the abdominal pain and duration of skin lesions (but do not prevent skin recurrence)
Refer to nephrology if evidence of renal involvement as treatment
They could consider ACEi, steroids or other treatments
URTICARIAL VASCULITIS
Clinical presentation resembling urticaria but see LCV on histopathology
Often idiopathic but can be associated with:
Autoimmune connective tissue disease: SLE, Sjogren’s, RA
Infections - HBV, HCV, EBV
Serum sickness
Medications - NSAIDs, fluoxetine, potassium iodide
Malignancies - usually haematological
Cutaneous features:
Unique from regular urticaria in that:
1. Urticarial papules and plaques last > 24 hours
2. More pain and burning than itch
3. May have residual haemorrhage
4. May esolve with postinflammatory hyperpigmentation
5. May have systemic symptoms (See below)
Rarely presents as: Bullae, EM like lesions, Livedo reticularis
Can be associated with or without angioedema
Urticarial vasculitis further broken down in to:
Normocomplementaemic (3/4 of cases)
Mostly skin limited
Average duration 3 years
Hypocomplementaemiac (1/4 of cases)
Hypocomplementaemic urticarial vasculitis is more likely to be associated with systemic changes with the following systems involved:
Arthralgia
Of hands, elbows, knees and ankles in 50% of patients with urticarial vasculitis
50% of patients with HUVS (see below) have frank arthritis
Pulmonary
Up to 20% have pulmonary symptoms - cough, laryngeal oedema, haemoptysis, asthma, COPD
The COPD is especially severe in smokers with urticarial vasculitis
GI
30% of patients
Abdominal pain, nausea, vomiting, diarrhoea
Renal
5-10% of patients with HUVS
Look for proteinuria/haematuria
Ocular
Uveitis/episcleritis occurs in HUVS
Hypocomplementaemic urticarial vasculitis syndrome (HUVS) is a more severe syndrome defined by specifc diagnostic criteria:

Patients with hypocomplementaemia who don’t meet the above criteria have hypocomplementaemic urticarial vasculitis but not HUVS
They are not thought to transition from one to another
Common abnormal labs urticarial vasculitis:
High ESR
Low C3, C4 and CH50 (this is decreased if components of classical complement pathway are low)
HUVS marked by:
Low serum complement
Plus presence of anti-C1q antibody and low C1q levels
HUVS and SLE:
HUVS can share features with SLE
Distinctive clinical findings in HUVS are:
Ocular inflammation (30%) - conjunctivitis, episcleritis, iritis,uveitis
Angioedema (>50%)
COPD like symptoms (50%)
Lab findings HUVS and SLE:
1/3 patients with SLE have anti-C1q antibodies
Up to 1/2 patients with HUVs have positive ANa
But patients with HUVS rarely have anti-dsDNA or anti-Sm antibodies
Diagnosis urticrial vasculitis:
H&E:
Finding of leucocytoclastic vasculitis is essential for diagnosis of urticarial vasculitis
(can often be subtle)
DIF:
70% Immunoglobulin, C3 or fibrin around blood vessels
Can see granular pattern along basement membrane in 80% of lesions - if this is accompanied by hypocomplementaemia this may be suggestive of a diagnosis of SLE
Treatment:
No RCTs
Antihistamines may reduce swelling and pain associated with lesions but do not alter course
Considerations:
Oral steroids effective but keep duration to a minimum
NSAIDs: indocmethacinDapsone
Colchicine
Hydroxychloroquine
MMF
Rituximab or IVIG may also be useful for recalcitrant hypocomplementaemic urticarial vasculitis
Schnitzler’s syndrome:
• Urticarial vasculitis
• Monoclonal IgM gammopathy
and at least two of the following:
Fever (periodic)
Arthralgia
Hepatosplenomegaly
Increased ESR
Increased WBC
Bone abnormality
Bone pain
Resolve with postinflammatory hyperpigmentation
Cryoglobulinaeimas:
Cryoglobulins are immunoglobulins that precipitate in the cold
Cryoglobulinaemia broken down in to 3 types:
Type 1:
Doesn’t cause a vasculitis
Usually due to monoclonal IgM (Less frequent IgG which is > than IgA)
Symptoms and signs due to ‘sludging’ of the blood vessels with microvascular occlusion

◦ Associated with plasma cell dyscrasias and lymphoproliferative disorders
◦ Present with:
‣ Livedo reticularis
‣ Raynaud’s
‣ Acrocyanosis
‣ Retiform Purpura
Type 2 and 3 - ‘mixed cyroglobulinaemias’
Approx 15% of patients with circulating mixed cryoglobulins present with symptoms due to cryoblobuniaemic vasculitis
Antibodies combine to form immune complexes which deposit in the blood vessels which activates complement causing a leucocytoclastic vasculitis
Causes a vasculitis that can involve both small and medium-sized vessels (preferentially small)
It typically affects:
Skin
Peripheral nervous system
Kidneys
Type 2 cryoglobulinaemias exhibit monoclonal IgM or IgG directed against polyclonal IgG

Type 3 cryobloulinaemia exhibit polyclonal IgM against polyclonal IgG

Occurs in setting of -
Infections:
Hepatitis C virus accounds for 70-90% of cases
>50% of patients with HCV infection have cryoglobulinaemia
Overt cryogloublinaemic vasculiltis develops in approx 5% of these cases
Higher prevalence in Southern Europe as increased prevalence HCV here
Hepatitis B virus (5% of cases)
EBV, CMV, Leishmania, Trepenoma
2. Connective tissue diease:
RA
Sjogrens
Systemic sclerosis
3. Lymphoproliferative disorder (5%)
B-cell non-hodgkin lymphoma
CLL
Macroglobulinaemia
Rarer: solid tumours (HCC)
Clinical cryoglobulinaemic vasculitis:
Cutaneous:
Commonest to have palpable purpura of lower extremities
Can see ecchymoses and dermal nodules
Rarer: urticaria, livedoe reticularis, necrosis, ulceration, bullae
Not typically cold induced (in contrast to vascular occlusive lesions in type 1 cryoglobulinaemia)
Arthritis - 70%
Peripheral neuropathy - 40% (typically sensory)
GI disease or hepatitis - 25%
Rarer:
Xerostomia/xerophthalmia
Endocrine issues (thyroid/gonads)
Investigations:
Elevated cryoglobulins
Can be falsely negative
Need to check during clinical flares and on more than one occasion
Blood sample should be kept warm at 37°C until it is spun in the lab to avoid a false negative
Check with lab their protocol prior to collecting cryoglobulins
Rheumatoid factor - is positive in 70-90% cases
Is an easier test to perform than cryogloublins
RF by definition is an autoantibody against the Fc portion of IgG
Fc portion is the bottom of the Y shape of the antibody
Types 2 and 3 mixed cryobloulinaemias consist of immune complexes containing antibodies to IgG so you often get positive RF
ANA - 20% positive
Complement - often low or undetectable C4
SPEP - 15% with mixed cryoglobulins have monoclonal gammopathy
Hepatitis and HIV serology
Pathology:
H&E:
Leucocytoclastic vasculitis
DIF:
Granular deposits of predominantly IgM and C3 in a vascular pattern observed in papillary dermis
Treatment:
Treat hepatitis C infection
Interferonα pluse ribavirin often can lead to resolution of cutaneous (100%), renal (50%) and neurological manifestations
I rare cases interferon can make the peripheral neuropathy worse
Considerations if severe:
Plasma exchange
Cyclophosphamide
Rituximab
ANCA associated vasculitis
Granulomatosis with polyangiitis (Wegener’s granulomatosis)
Microscopic polyangiitis
Eosinophilic granulomatosis with polyangiitis (Churg Strauss)
Together these conditions have annual incidence of 20 cases per million people in N. America and Europe
Anti-neutrophil cytoplasmic antibodies (ANCA) refer to IgG auto-antibodies that target antigens in the cytoplasm of neutrophils (mostly) and monocytes
To improve specificity two tests are usually done on a patients blood:
Indirect immunoflourescence on a patient’s neutrophils can demonstrate two vasculitis relevant patterns of staining:
cytoplasmic: c-ANCA
perinuclear: pANCA
ELISA which can specifically detect antibodies to the relevant antigens
Proteinase 3 - anti-PR3 antibodies
Myeloperoxidase - anti-MPO antibodies
ELISA can detect the amount of antibody present as a titre
The higher the titre, the more antibody there is (ie 1:64 > 1:8)
About 85% of patients with cANCA pattern have PR3 antibodies
About 90% of patients with pANCA pattern have MPO antibodies
Often do IIF first which detects pANCA or cANCA staining and if positive you then do ELISA to specifically detect anti-PR3 antibodes or anti-MPO antibodies
However, up to 5% of serum samples are positive for ELISA only so if there is a high index of suspicion for ANCA associated vasculitis should perform ELISA even if IIF is negative
Anti-PR3 ANCA:
+ve in up to 90% of patients with GPA (Wegener’s)
+ve in 30% of patients with MPA
Anti-MPO ANCA:
+ve in 60% of patients with MPA
+ve in 60% of patients with EGPA (Churg-Strauss)
Can use ANCA for disease monitoring and also for checking response to treatment
Presence of ANCA within first year following remission has been associated with disease relapse
Pathogenesis:
PR3 and MPO are intrecellular neutrophillic proteins that can translocate to the neutrophil cell surface after activation by cytokines (eg TNFα)
An ANCA antibody can then bind to these antigens resulting in adhesion of neutrophils to the blood vessel endothelium
This results in release of inflammatory mediators, vessel damage and recruitment of additional inflammatory cells
As there is a lack of immune complex deposition on immunoflourescence we call this pauci-immune
Prognosis:
Morbiditiy and mortality of ANCA-associated vasculidities are relatively high due to:
Systemic manifestations disease
Complications of immunosuppressive therapy
Poor prognostic factors:
Delay in diagnosis
Renal impairment
Propensity for relapse
Older age
Presence of ANCA
Workup for systemic vasculitis:
Do extensive history for past and present clinical features (including)
Once a diagnosis is made the severity of the disease should then be assessed
Is it limited: eg chronic ENT disease
Is it severe (organ or life threatening): renal, pulmonary or neurological disease
Treatment is aimed at:
Remission induction
To switch off the vasculitis activity
Done over a course of 3-6 months
Remission maintenance
To keep disease under control
The treatments for the various ANCA vasculidities in general are quite similar
GRANULOMATOSIS WITH POLYANGIITIS (formerly Wegener's granulomatosis)
Chronic systemic vasculitis
Small and medium sized arteries affected
Classic triad of symptoms for granulomatosis with polyangiitis:
1. Upper respiratory disease
2. Lower respiratory disease
3. Renal disease
Clinical findings:
Peak incidence age 30-50
Can involve multiple organ systems
MICROSCOPIC POLYANGIITIS
Can have quite similar clinical findings to GPA but without:
The granulomas
The upper respiratory findings
The progressive clinical course of MPA typically leads to renal failure and/or pulmonary haemorrhage
Antibodies:
Anti-MPO antibodies +ve 60% of patients
Anti-PR3 antibodies +ve 30% of patients
Skin biopsy:
Segmental necrotising vasculitis of small blood vessels and to lesser extent medium-sized arteries
No evidence of granulomatous inflammation
Treatment:
2 phases:
Induction of remission
Maintenance therapy
Induction remission:
Steroids initially (1mg/kg/day pred)
Addition of cyclophsphamide with significant organ involvement (renal, pulmonary, neurologic)
Can give cyclophosphamide for 6 months (orally 2mg/kg/day or IV Pulses 0.5-1g/m2/month)
Pulse therapy reduces total drug dose and incidence of side effects (eg bladder cancer)
Recent data suggest corticosteroids plus rituximab may be equally effective as steroids/cyclophosphamide
Maintenance of remission:
Methotrexate
Azathioprine
MMF
IVIG
Plasma exchange can be considered in ANCA-positive patients
Biologics (eg infliximab) has shown promise but further evidence required
Prognosis:
Higher rate of relapse compared to classic PAN (regardless of severity)
Lower rate than those with Wegener’s
Persistence of ANCA despite induction of remission associated with increased risk of relapse
EOSINOPHLIC GRANULTOMATOSIS WITH POLYANGIITIS (formerly Churg-Strauss syndrome)
Granulomatous small vessel vasculitis affecting mainly blood vessels of:
Lungs (severe asthma, allergic rhinitis)
GI tract
Peripheral nerves
Lesser extent:
Skin
Heart (leading cause of death)
3 phases:
1st phase - symptoms of allergic rhinitis, nasal polyps and asthma persisting for years
2nd phase - peripheral eosinphilia, respiratory tract infections, GI symptoms
3rd phase - Systemic necrotizing vasculitis with granulomatous inflamamtion which can occur several years to decades after initial symptoms
Investigations:
Elevated IgE
p-ANCA
Anti-myeloperoxidase +ve in 60%
POLYARTERITIS NODOSA
Necrotising vasculitis of medium sized muscular arteries
Comes in 2 presentations:
1. Classic systemic form
2. Cutaneous PAN (limited systemic involvement)
PAN can affect multiple organs but tends to spare one organ: the lung
Clinical:
Skin changes:
Palpable purpura lower legs
Painful subcut nodules following course of blood vessels
Lacy livedo reticularis interspersed amongst these purpura and nodules
PAN can affect multiple organs but tends to spare one organ - the lung
Kidney damage is due to issues with perfusion
Don’t get a glomerulonephritis in PAN
PAN associations:
• Hepatitis B > C
• HIV
• CMV
• Streptococcal infections
• IBD
Diagnosis (systemic PAN)
3 out of 10 criteria put out by ACR
Think of signs and symptoms first and then workup:
Investigations:
Blood pressure
Urinalysis
FBC (anaemia)
U&E
LFT
ESR/CRP
Hepatitis and HIV serology
ANCA (should be negative)
ASOT
Biopsy findings PAN:
LCV along with the arteritis of medium sized arterieis in the deep dermis and subcut tissue
May see a lobular panniculitis next to the involved vessels
Imaging:
• Renal angiogram to look for aneurysms or renal artery stenosis
Treatment:
• Immunosuppression:
◦ Systemic steroid for at least 6 months along with MTX or cyclophosphamide
• Organ specific management should involve other consultants (eg cardiology, nephrology)
• Treat any underlying causes (eg strep and Hep B)
KAWASAKI DISEASE
An acute febrile illness with vasculitis affecting the small and medium-sized vessels throughout the body, in particular the coronary arteries
Diagnostic criteria:
• Need fever of at least 5 days
and 4 out of 5 of other diagnostic criteria (Think CRASH and BURN)
• Burn= burning hot fevers > 39 celsius for five more days
Then four out of five of:
Conjunctivitis
Rash: polymorphous exanthem which can be peri-anal
Adenopathy: Cervical lymphadenopathy
Strawberry tongue or other oral changes
Hands/feet: erythema, oedema, eventual desqumation
Can also have:
Irritability, malaise, arthralgias
Uveitis
Gastroenteritis
Urethritis that may be symptomatic
Most cases are in children less than 5 years old
Especially around 10 months of age
Most patients have asian ancestry, especially Japanese kids
Coronary artery aneurysms can occur several weeks after symptom onset in about 25% of untreated children
These aneurysms are due to a medium vessel vasculitis
Lab investigation suspected Kawasaki disease:
FBC: WCC elevated, anaemia, thrombocytosis or thrombocytopaenia
U&E
LFT: Hypoalbuminaemia
CRP/ESR elevation
Nasal and throat swab looking for streptococcus
ASOT (As children with scarlet fever can have similar findings with fever, exanthem and strawberry tongue)
Blood culture
Urinalysis
Urgent ECHO looking for cardiac invovlement
Treatment:
Acutely ill:
IVIG 2g/kg over 12 hours as a single dose
Check IgA levels before to outrule IgA deficiency
If give IVIG to IgA deficienct patient they will see the IgA in IVIG as foreign and mount an immune reponse to it that can lead to anaphylaxis
High dose aspirin: 80-100mg/kg/day
This is the only time aspirin is acceptable to give to children
If patients fail to respond to IVIG and aspirin they may need:
Repeat IVIG treatment
Corticosteroids
Steroid sparing agents (ciclosporin or cyclophosphamide)
Behcet's disease
Inflammatory disease charachterized by recurrent oral apthous ulcers and numerous potential systemic manifestations
Many, but not all, clinical manifestations of Bechet disease are believed to be due to vasculitis
Among the systemic vasculitides, Behcets disease remarkable for ability to involve blood vessels of all sizes (small, medium and large) on both the arterial and venous sides of the circulation
Epidemiology:
Generally equal sex ratio
Affects young adults (20-40)
More common (and often more severe) along the ancient silk road from Eastern Asia to Mediterranean
Highest prevalence in Turkey (80-370 cases per 100,000)
US/Northern Europe (0.12-7.5 per 100,000)
Close relationship in geographic distribution of HLA-B51 and prevalence of BD
Odds ratio of developing BD with HLA-B51 +ve: 5.78
Aetiology and pathogenesis:
Underlying cause of Behcet syndrome unknown
As with other autoimmune diseases:
May represent aberrant immune activity triggered by exposure to an agent, perhaps infectious, in patients with a genetic predisposition to the disease
Genetic: HLA genes
Environmental influences: ? Abnormal immune response to microbes (eg Streptococcus or H pylori)
Clinical:
Mucocutaneous
Oral apthous ulcers
Painful, frequent, extensive
Urogential ulcers
Most specific lesion, less frequent, scar
Cutaneous (up to 75% of patients)
Acneiform lesions
Erythema nodosum
Pyoderma gangrenosum
Ocular (25-75%)
Anterior/posterior/pan uveitis
Retinal vasculitis
Optic neuritis
Neurologic disease (<10%, late onset, highly variable)
Parenchymal disease (brainstem, cerebrum, spinal cord)- wide range symptoms/signs
Non-parenchymal disease (meningism, cerebral venous thrombosis)
Vascular disease
Pulmonary artery aneurysm (Mortality approximately 25%) and thrombosis
Venous thrombosis (DVT, Budd-Chiari, Dural sinus thrombosis, SVC/IVC thrombosis)
Estimated 14-fold increased risk of venous thrombosis compared to controls
Arthritis
Non-erosive, aysmmetric, usually non-deforming
Gastro-intestinal
Abdominal pain, diarrhoea, bleeding (similar to IBD)
Renal and cardiac involvement
Infrequent but can occur
Pathergy:
Associated with Behcet’s disease
Is the development of cutaneous papulopustular lesions 24 hours after cutaneous trauma
More frequently positive in Behcet’s disease patients from endemic areas (50-75%) then non-endemic areas such as the US (10-20%)
Causes pathergy: Behcet’s, Pyoderma gangrenosum, Sweet’s syndrome
Diagnosis:
Behcet syndrome is best diagnosed in the context of recurrent apthous ulcerations along with characteristic systemic manifestations
There are no pathognomonic laboratory tests
Therefore diagnosis is made on the basis of clinical findings
Multiple diagnostic criteria exist:
One such example is the International Study Group Diagnostic Criteria (1990)
Another example is the International Criteria for Behcet’s Disease (ICBD, 2006)
Each of several findings are assigned a point value
Require at least 3 for diagnosis of Behcet’s