
URTICARIA INTRODUCTION
Urticaria (commonly known as 'hives') is one of the most frequently encountered conditions in both general practice and dermatology, affecting up to 20% of the population at some point in their lives. It presents as an intensely pruritic rash characterised by transient weals that typically resolve within 24 hours, often accompanied by a surrounding erythematous flare.
'Wheal and flare' reaction:
The wheal refers to the raised, pale, oedematous centre — caused by increased vascular permeability allowing fluid to escape into the superficial (papillary) dermis
The flare is the surrounding erythematous zone — caused by reflex vasodilation
The transient nature of individual lesions (resolving within 24 hours) is a defining clinical feature that helps distinguish urticaria from other dermatoses.
It can be classified in different ways:
Based on pathogenesis:
Immunologic or non-immunologic
Based on length of symptoms:
Acute urticaria: Symptoms last < 6 weeks
Chronic urticaria: > 6 weeks of daily or almost daily wheals
Episodic: Acute intermittent or recurrent
Based on if symptoms and signs are inducible or non-inducible (also known as ‘spontaneous’)
Lifetime prevalence for all types of urticaria is 8 to 24%
Chronic urticaria lifetime prevalence is 1-2%


Angioedema:
Is the result of deeper swelling in the reticular dermis and subcutaneous tissue/submucosa (compared to weals affecting the more superficial papillary dermis)
Common sites are the eyes, lips, oropharynx and genitals but can occur in other sites like the gastrointestinal tract and upper airways
Angioedematous swellings last longer then weals and can persist for a few days
Weals and angioedema often coexist, but either can occur separately

Some forms of urticaria can be associated with systemic symptoms such as:
Lethargy, malaise
Arthralgia
GI disturbance
Wheeze and/or mucosal involvement
Acute urticaria can be a presenting sign of anaphylaxis
PATHOGENESIS
The mast cell is a cell that can be found predominantly in skin, submucosa and bowel mucosa
Histamine is produced and stored in the mast cell in granules and is the most important mediator of urticaria
Urticaria results from mast cell (+/- basophil) degranulation which leads to the release of preformed vasoactive mediators (eg histamine and others such as tryptase)
Get symptoms and signs due to the resulting vasodilation, increased blood flow and increased vascular permeability
Following the initial release of preformed mediators mast cells and other cells (eg eosinophils, neutrophils) make and release secondary mediators like prostaglandins (cyclo-oxygenase enzyme pathway) and leukotrines (lipoxygenase pathway) which are derived from arachidonic acid and further contribute to the symptoms and signs.
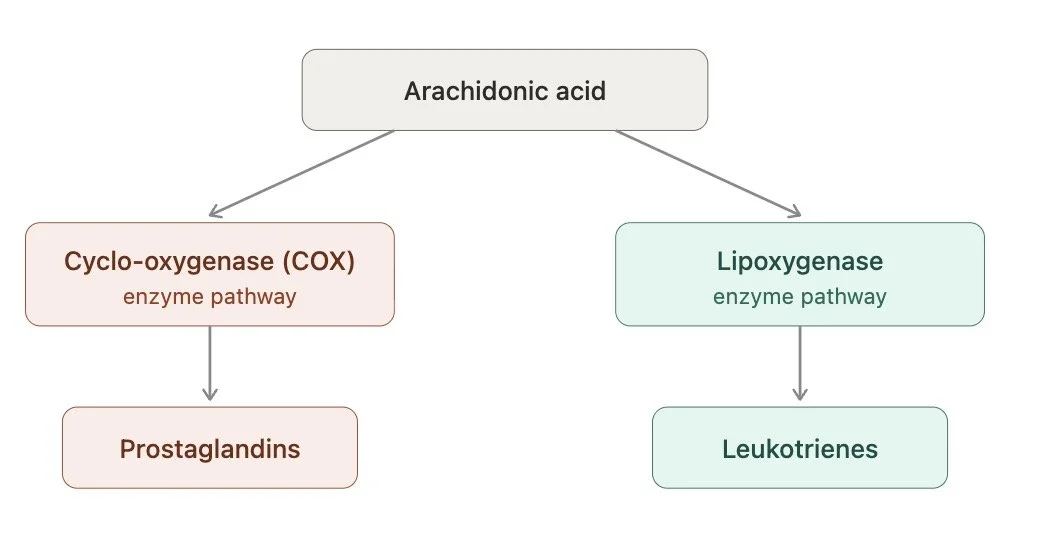
Angioedema can involve additional mechanisms such as bradykinin release and as such antihistamines may not be as effective
CLASSIFICATION BASED ON PATHOGENESIS
There are a variety of immunologic, non immunologic, physical and chemical stimuli that can cause urticaria
Angioedema without weals can be seen in conditions where bradykinin is increased
IMMUNOLOGICAL
Going into the immunological mechanisms in more detail, this can be related to the adaptive (allergic, autoimmune, immune complex formation) or the innate immune system (activation through toll like receptors or autoinflammatory syndromes)
Allergic (type 1 hypersensitivity reaction)
This is the classic allergic reaction which can cause an acute urticaria
Mast cells express high affinity IgE receptors (FcεRI) on their surface. IgE antibodies sit on these receptors, each with a specific antigen-binding site directed against a particular allergen
When the relevant allergen is encountered, it binds to and bridges two adjacent IgE molecules on the mast cell surface — this is called cross-linking. Cross-linking of IgE triggers activation of the mast cell, leading to degranulation and release of histamine and other preformed mediators, causing urticaria
Types of allergens that can cross-link mast cell-bound IgE include foods, drugs, inhalants, and allergens from insect bites and stings
Autoimmune
Generally chronic spontaneous urticaria is the most likely urticarial condition you will encounter in the Dermatology clinic
Chronic spontaneous urticaria is now increasingly recognised as an autoimmune condition
Over the past 10 years, the understanding of the immunological mechanisms has evolved significantly
CSU can be now classified into two main autoimmune endotypes
Understanding these endotypes is clinically important as they have distinct biomarker profiles and may predict response to different treatments
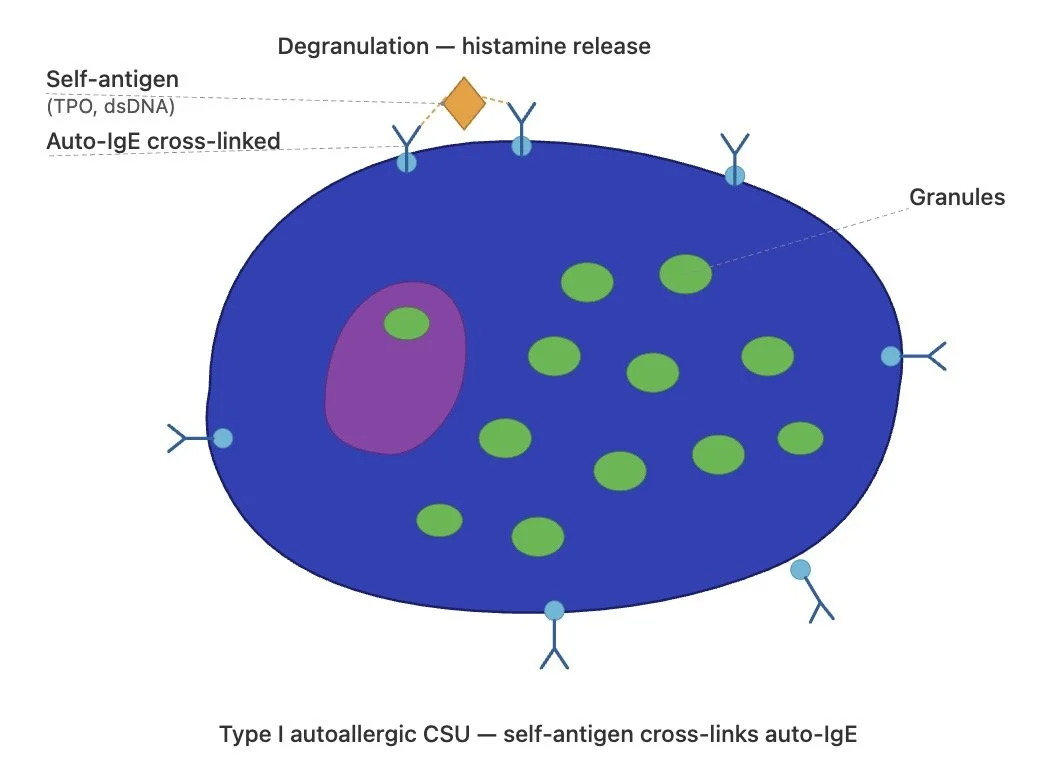
Type I autoallergic CSU (autoallergy)
This is the commonest endotype
In type I autoallergic urticaria, patients produce auto-IgE antibodies directed against self-antigens
These internal antigens include thyroid peroxidase (TPO), double-stranded DNA (anti-dsDNA) and others
These auto-IgE antibodies bind to the high-affinity IgE receptor (FcεRI) on mast cells and when cross-linked by the relevant autoantigen, the mast cell degranulates and releases histamine, causing wheals
This is called an ‘autoallergic’ condition because the mechanism mirrors a classical type I hypersensitivity reaction, but the allergen is an internal self-antigen rather than an external allergen
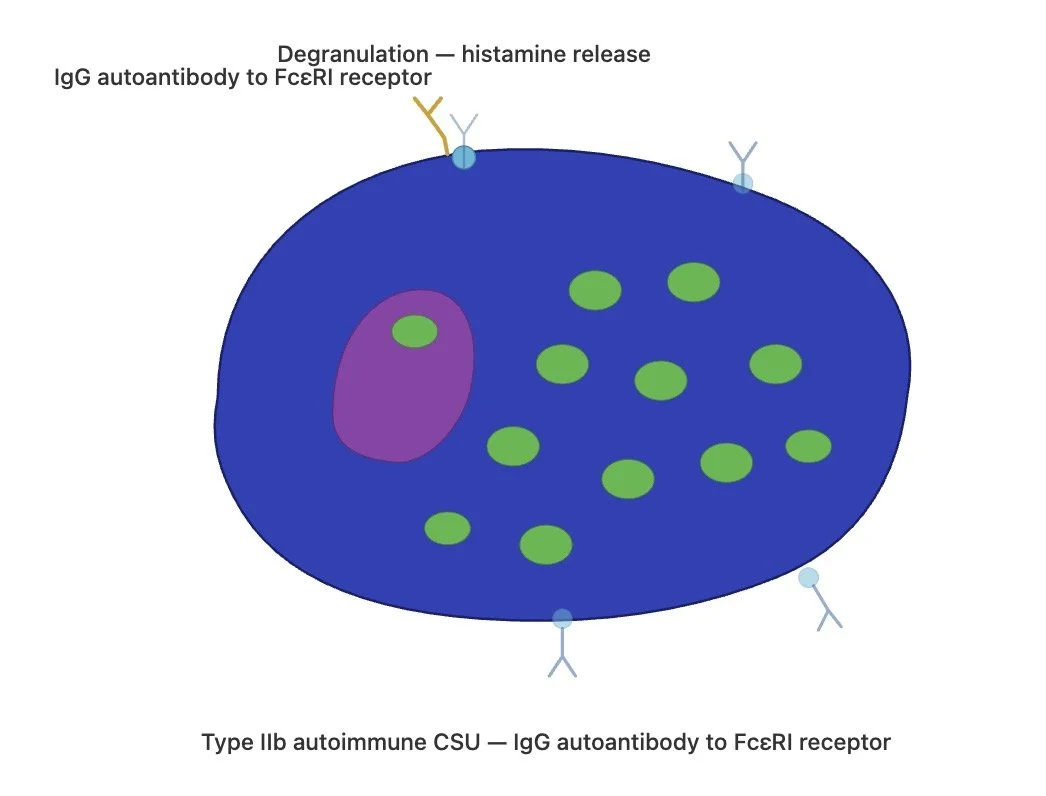
Type IIb autoimmune urticaria
In type IIb autoimmune urticaria, patients produce IgG autoantibodies directed against high-affinity IgE receptor (FcεRI) itself or against IgE bound to the receptor. These IgG autoantibodies activate mast cells, basophils and eosinophils, leading to degranulation and histamine release.
This is classified as type IIb (rather than IIa) because the autoantibodies modify the function of the cell (stimulating degranulation) rather than destroying the cell.
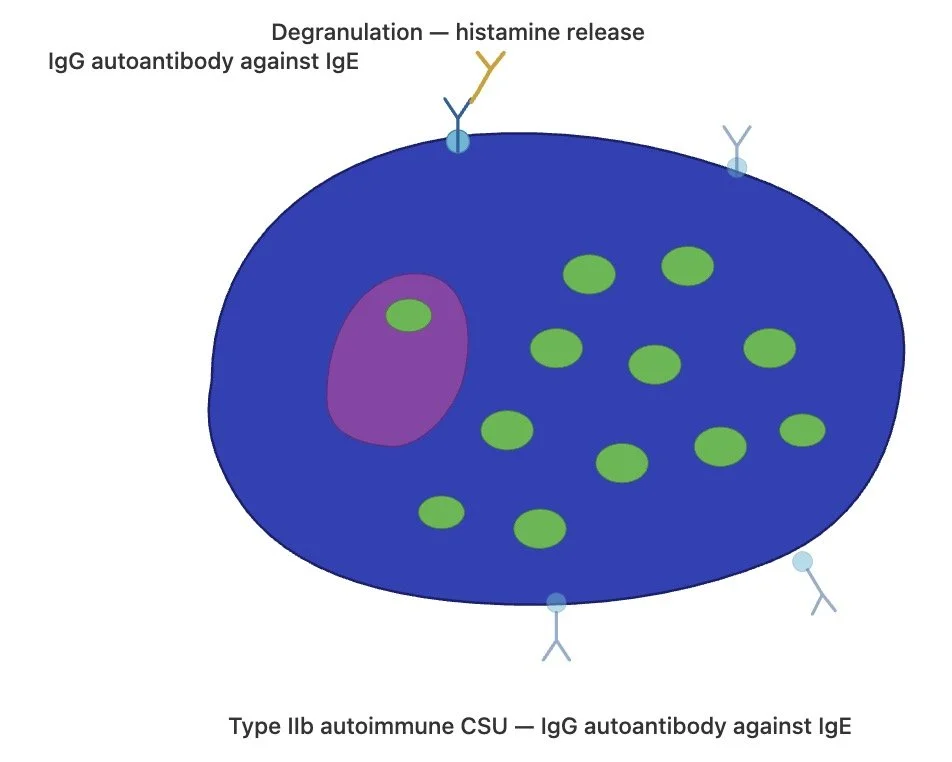
While the mast cell is the primary effector cell in all forms of urticaria, in type IIb autoimmune CSU the IgG autoantibodies also activate basophils and eosinophils. This is because basophils, like mast cells, express FcεRI on their surface and can be targeted by the same IgG autoantibodies
The BAT and BHRA are laboratory tests that detect this basophil activation — a positive result supports a type IIb endotype, while a negative result is more in keeping with type I autoallergic disease
It is important to note that in clinical practice many patients fall somewhere between these two endotypes and it may note even be able to classify them into one category
The endotypes represent a spectrum rather than two entirely discrete categories
Immune complex formation and C5a
This pathway is seen in conditions where circulating immune complexes are present, such as hepatitis B and C, EBV infection, SLE, and serum sickness.
Immune complexes activate the complement cascade and this generates C5a (a small protein fragment called anaphyloxin)
C5a binds to C5a receptors on mast cells, causing degranulation and histamine release without any IgE invovlement
Because mast cell activation occurs through C5aR rather than the IgE/FcεRI pathway, these conditions are often resistant to standard urticaria treatment, including high-dose H1 antihistamines and omalizumab
The same mechanism underlies urticarial vasculitis, which represents the more severe end of the spectrum where neutrophil recruitment additionally causes leukocytoclastic damage to vessel walls
Innate immune system:
Toll-like receptors:
Mast cells can also be activated through Toll like receptors
These TLRs recognise pathogen-associated molecular patterns (PAMPs) from microbes which may explain why viral infections and other infections can trigger or exacerbate urticaria.
Auto-inflammatory syndromes:
Get dysregulation of innate immunity leading to persistent uncontrolled inflammation in the absence of an identifiable trigger
Get excessive cytokine production which can lead to urticaria
Urticaria-like rashes can be feature of autoiflammatory syndromes such as:
Shnitzler syndrome
Cryoprin-associated periodic syndromes (CAPS)
Adult-onset Still’s disease
NON-IMMUNOLOGICAL
Mast cells can also be activated through non-immunologic mechanisms, where degranulation occurs without involvement of the adaptive or innate immune system
Direct activation through MRGPRX2 receptors
MRGPRX2 is a receptor expressed on mast cells that can be directly activated by a range of drugs and neuropeptides, causing degranulation independently of IgE.
Drugs that activate mast cells through MRGPRX2 include opioids (e.g. codeine, morphine), radiocontrast media, fluoroquinolones and vancomycin.
Substance P, a neuropeptide involved in pain and inflammation signalling, also activates this receptor.
Approximately 10% of patients with urticaria may have gain-of-function mutations in MRGPRX2, leading to more severe disease.
Clinical trials targeting this receptor are currently underway.
CLINICAL CLASSIFICATION
ACUTE URTICARIA
Episodes last < 6 weeks
75% of urticaria cases are acute
Angioedema more common in acute cases
No cause found in over 50% with acute urticaria
Causes include:
Medications- antibiotics, NSAIDs, anticonvulsants, codeine
Infections (particularly viruses in kids) (think hepatitis in adults)
Foods- nuts, shellfish, eggs, cow's milk
Insect bites/stings
In acute urticaria always ask about angioedema.
In particular ask about throat swelling, wheeze/hoarseness, GI side effects.
If have worrying symptoms consider giving subcut adrenaline and send to hospital
If just have urticaria- give non sedating anti-histamines as first line
CHRONIC URTICARIA
Urticaria lasting > 6 weeks
CHRONIC SPONTANEOUS URTICARIA
This is the commonest type of chronic urticaria where no clear cause is found for it
CSU has a tendency toward spontaneous resolution even without treatment, but the duration varies significantly between individuals
Predictors of longer disease duration:
Female sex
Older age at onset
Angioedema (as a component of CSU)
Autoimmune markers (especially thyroid autoimmunity)
Severe baseline activity
Co-morbid inducible urticaria
Systemic inflammation
Relapse risk:
Even after resolution, CSU can relapse. A relapse risk of 13-75% has been reported in the literature. The longer the patient has had the condition, the more likely it is to relapse
Associations:
INDUCIBLE URTICARIAS
Usually chronic
Weals reproducibly induced by same physical stimulus
Usually appear within few minutes of stimulus and last < 2 hours
(except delayed pressure urticaria which can take 30 min to 12 hours to develop and lasts for a few days)
Some inducible urticarias can present as a spectrum of symptoms from simple pruritus through to severe symptoms
Tests can be confirmed on provocation testing if have this set up
Please note that cold urticaria in particular warrants close attention as it can cause anaphlaxis (eg if person goes cold water swimming or jumps in a plunge pool)
Dermographism (symptomatic): The commonest inducible urticaria. Wheals occur at sites of minor trauma such as scratching or friction.
Provocation test - Stroke the skin with a tongue depressor and wait 10 minutes

Dermographism- whealing induced by stroking the skin
Cold contact urticaria: Triggered by exposure to cold water, cold wind or cold surfaces. This is the inducible urticaria most associated with anaphylaxis — particularly with large surface area cold exposure such as swimming in cold water
Patients should carry an adrenaline auto-injector
Consider checking cryoglobulins
Provocation test - place ice cube in sealed plastic bag over forearm for up to 10 mins and allow to rewarm
Cholinergic urticaria: Triggered by a rise in core body temperature, typically through exercise or emotional stress
Characteristically produces small (2–4mm) monomorphic papular wheals rather than the larger wheals seen in other forms of urticaria
Provocation test - exercise patient in warm environment
Delayed pressure urticaria:
Unlike other inducible urticarias, symptoms are delayed — onset 30 minutes to 12 hours after sustained pressure, and swelling can last for days
Occurs at sites of prolonged pressure such as the soles of the feet after walking, the buttocks after sitting, or under tight waistbands
Can be difficult to treat
Solar urticaria:
Triggered by UV exposure (and sometimes visible light)
Consider checking ANA and porphyrins
May benefit from referral to a specialist centre for phototesting to identify the action spectrum, which can guide UV prophylactic phototherapy
Aquagenic urticaria:
Triggered by contact with water at any temperature
Annual FBC as can be associated with PRV, other haem disorders
Provocation test - wet towel for few minutes over most affected area or immerse body part in water (at 37 degrees)
Heat contact urticaria:
Triggered by direct contact with warm objects or hot water. Much rarer than cold contact urticaria.
Vibratory urticaria:
Triggered by vibratory stimuli such as power tools, lawnmowers or motorcycles.
Exercise-induced anaphylaxis: Anaphylaxis triggered by exercise, sometimes only in combination with a co-factor such as food ingestion (particularly wheat/omega-5 gliadin) or NSAIDs
Distinct from cholinergic urticaria in that it can progress to full anaphylaxis
MANAGEMENT URTICARIA INTRODUCTION
History and exam is key
Assess impact on quality of life (eg DLQI)
Activity of disease should be measured if considering a second line treatment (eg UAS 7 score)
The UAS 7 score ask the patient to score their severity of urticaria based on the number of weals (mild, moderate, severe) and pruritus (mild, moderate, severe) every day over a period of 7 days
Ask about sleep and work/school disturbance
Enquire about symptoms/signs to suggest a vasculitic process:
Are the lesions persistent rather than self-limiting?
Are the lesions tender and painful rather than itchy?
Does the skin show evidence of residual petechiae, purpura or bruising
Does the patient have any symptoms/signs of underlying disease - fever, malaise, arthralgia, hypertension, blood/protein in urine
Enquire about any potential triggers and associations
TRIGGERS AND ASSOCIATIONS
Triggers:
Stress- stressful events may precede urticaria
Infections:
Viral infections
Chronic infections- if have found no cause for chronic urticaria enquire about chronic dental infections, reflux (H pylori), recurrent UTIs, sinusitis
Medications - NSAIDs, Opioids
Heat, exercise
Tightly fitting garments
Of note:
Patients often enquire about food as a cause of chronic urticaria but a history enables mediated food allergy to be ruled out pretty quickly
In IgE mediated food allergy symptoms typically occur within 60 minutes of exposure to the offending food rather than coming on overnight or being present first thin in the morning
Also urticaria and angioedema associated with food allergic reactions rarely occur in isolation and you usually have additional symptoms: eg oropharyngeal itch, wheeze, vomiting, abdominal pain
Caveat - There are very rare trickier exceptions:
Food dependent exercise induced anaphylaxis:
The "Two-Hit" Trigger: An allergic reaction which only occurs when a specific food is consumed and then followed by a secondary cofactor—usually physical exertion, but sometimes alcohol or NSAIDs—within a few hours.
The Common Culprits: The most frequent offenders are Omega-5 gliadin (a protein in wheat), Lipid Transfer Proteins (tough defensive proteins in plant and fruit skins), and crustacea (shellfish like prawns).
Why it mimics chronic urticaria: Because the patient usually eats the trigger food frequently without any issue while resting, the hives seem entirely random and inconsistent, hiding the true cause.
Alpha-Gal Syndrome (Red Meat Allergy)
The Culprit: A unique allergy to a carbohydrate molecule (alpha-gal) found in the meat of mammals (beef, pork, lamb), which is typically acquired after a person is bitten by a tick.
The Delayed Reaction: Unlike typical protein-based allergies that strike within an hour, the body takes much longer to digest complex carbohydrates, meaning symptoms do not appear until 3 to 8 hours after eating the meat.
Why it mimics chronic urticaria: Because of the massive time delay, a patient might eat a burger for dinner and wake up at 3:00 AM covered in hives, making it appear as though the urticaria flared up spontaneously overnight.
INVESTIGATIONS
Acute/episodic spontaneous urticaria:
Skin prick or RAST testing may help confirm type 1 hypersensitivity reaction if suspected but don’t use these tests in chronic urticaria
FBC, CRP/ESR may help identify infective cause
Chronic urticaria:
There are no diagnostic tests for CSU
Could consider:
FBC
ESR/CRP (if elevated could consider urticarial vasculitis, autoinflammatory disease)
IgE
TFT and Thyroid antibodies (association autoimmune thyroid disease)
20% patients have thyroid antibodies in CSU vs 6% normal population
Other considerations
If GI symptoms consider TTG antibodies and exclusion of H Pylori
Dentist appointment if any concerns about dentition (? Abscess)
Autoimmune disease (eg coeliac disease): clinical suspicion should remain high but screening in absence of associated features generally not suggested
If concerned for an urticarial vasculitis may consider a biopsy and do a vasculitic screen including complement levels
Biopsy:
Usually don’t require in standard urticaria but may perform in unusual patterns or in cases of suspected vasculitis
Biopsy of an uncomplicated urticarial wheal is often subtle and findings can be quite underwhelming. You may see:
Dermal oedema — separation of collagen bundles in the upper and mid dermis, often the most striking feature
A sparse perivascular infiltrate — predominantly lymphocytes, with variable numbers of eosinophils, neutrophils, and occasional basophils
Mild vascular dilatation — without vessel wall damage
No leukocytoclasia, no fibrinoid necrosis, no red cell extravasation, and no vasculitis
The epidermis is normal (helpful in distinguishing from eczematous or other inflammatory dermatoses)
FIRST LINE TREATMENT CSU - 2ND GENERATION ANTIHISTAMINES
First-Line Treatment & Administration
Choose a modern 2nd-generation H1-antihistamine: Standard first-line options include fexofenadine, bilastine, cetirizine, levocetirizine, desloratadine, loratadine.
Dose regularly, not PRN: To effectively prevent wheals and angioedema, these medications must be taken on a regular, daily schedule rather than on an "as-needed" basis.
Escalation & Adjustments
Uptitrate for poor control: If standard doses do not provide sufficient improvement, increase the dose up to fourfold. Uptitration should be done every 2–4 weeks until symptoms are controlled.
Switch, don't mix: If a patient has an inadequate response or poor tolerance to their first medication, consider switching them to a different 2nd-generation H1-antihistamine.
European guidelines advise against combining two different 2nd-generation H1-antihistamines simultaneously due to a lack of supporting evidence.
Maintenance & Tapering
Maintain the regimen: Encourage patients to continue their daily antihistamines for a set period, even if they have been lesion-free.
Taper gradually: Once complete symptom control is achieved and sustained, attempt a stepwise dose reduction (eg decrease by a tablet every month if remain symptom free)
| Medication | Dosage / Frequency |
|---|---|
| Bilastine | 20 mg od |
| Cetirizine | 10 mg od |
| Desloratadine | 5 mg od |
| Fexofenadine | 180 mg od |
| Levocetirizine | 5 mg od |
| Loratadine | 10 mg od |
Antihistamines in paediatric population (adapted from BAD guidelines 2021)
Preferred first-line options for children include cetirizine, desloratadine, loratadine, and fexofenadine
Many are licensed for ages 2–11 but ‘when needed, these antihistamines can also be used below age 2 years, despite the licensing age cutoff, given their overall good safety profile’
Administer on a regular, daily schedule using licensed doses based on age and weight
As with adults if symptoms are poorly controlled, carefully increase the frequency up to a maximum of four times a day, adjusting strictly for the child's age and weight
Again its recommended not to combine multiple 2nd generation H1 antihistamines
| Drug | 1 month – 1 year |
1–2 years | 2–5 years | 6–11 years | 12–17 years | Adults (≥18 years) |
|---|---|---|---|---|---|---|
| Cetirizine Liquid 5 mg/5 mL; Tabs 10 mg | ✕ | 250 mcg/kg bd ¹ | 2.5 mg bd | 5 mg bd | 10 mg od | 10 mg od |
| Levocetirizine Tabs 5 mg | ✕ | ✕ | ✕ | 5 mg od | 5 mg od | 5 mg od |
| Fexofenadine Tabs 30/120/180 mg | ✕ | ✕ | ✕ | 30 mg bd ¹ | 180 mg od | 180 mg od |
| Loratadine Liquid 5 mg/5 mL; Tabs 10 mg | ✕ | ✕ | <31 kg: 5 mg od | <31 kg: 5 mg od ≥31 kg: 10 mg od |
10 mg od | 10 mg od |
| Desloratadine Liquid 2.5 mg/5 mL; Tabs 5 mg | ✕ | 1.25 mg od | 1.25 mg od | 2.5 mg od | 5 mg od | 5 mg od |
¹ Unlicensed use for this age group. Cetirizine unlicensed under 2 years. Fexofenadine licensed for urticaria from age 12 (may be used off-label from 6 years).
Antihistamine use in pregnancy
Initial Management Strategy -
Safety and general principles: Approximately 15% of pregnant women use antihistamines, and there have been no reported birth defects associated with 2nd-generation H1-antihistamines. Their use—even during the first trimester—is not associated with an increased risk of major malformations
Start with standard doses: The initial management strategy is the same as for the general population: start treatment with standard doses of modern 2nd-generation (non-sedative) H1-antihistamines
Preferred options (medications of choice): Loratadine and cetirizine are the preferred antihistamines based on safety data and current guidelines
Updosing considerations (pregnancy):
If standard doses are insufficient, consider uptitrating the anti-histamines
However, because the safety of these higher-than-approved doses has not been specifically evaluated in pregnant patients, the potential risks and benefits must be discussed with the patient
During Lactation (Breastfeeding)
Safety and general principles: All H1-antihistamines are excreted into breast milk in low concentrations. 2nd-generation options are advised because their transfer rate to breast milk is minimal and they prevent the infant from suffering adverse side effects.
Preferred options: Cetirizine, loratadine, and fexofenadine are the most thoroughly studied antihistamines for nursing mothers.
Updosing considerations (lactation): In refractory (difficult-to-treat) cases, higher doses of 2nd-generation antihistamines might be safely used while nursing, as studies on higher doses of loratadine show minimal transmission into milk.
What to Strictly Avoid
1st-generation H1-antihistamines: These should be avoided entirely for both pregnant and lactating women. If taken while breastfeeding, they are transmitted through breast milk and can cause irritability and drowsiness (sedation) in nursing infants.
MONTELUKAST
Consider montelukast 10mg nocte in addition to a second-generation H1-antihistamine in people whose symptoms are not controlled
Montelukast may be more beneficial in cases where it is exacerbated by aspirin, NSAIDs or in those with delayed pressure urticaria or where angio-oedema is the predominant symptom
Neuro-psychiatric effects can occur in a minority of people, particularly children so counsel parents about this low risk
When prescribing, you must counsel parents and patients to remain highly vigilant for symptoms such as dysphemia (stuttering), nightmares, night terrors, aggression, and sudden behavioural changes
1ST GENERATION ANTIHISTAMINES
| Medication | Dosage / Frequency |
|---|---|
| Chlorphenamine (Piriton) | 4 mg every 4–6 hours (max 24 mg/day; elderly max 12 mg/day) |
| Hydroxyzine (Atarax) | Initially 10–25 mg nocte Max 25 mg tds–qds (elderly max 25 mg bd) |
1st generation anti-histamines should not be offered routinely unless there is no alternative due to concerns about short term and long term effects on CNS (sedation, psychomotor impairment and REM sleep disturbance)
This is particularly relevant in the paediatric population
Do not up dose first generation anti-histamines
H2 ANTIHISTAMINES
H2-antihistamines (eg famotidine) are no longer recommended as a standard step in the European urticaria treatment algorithm due to a lack of strong clinical evidence
The current guidelines make no official recommendation for or against combining H1- and H2-antihistamines for patients who do not respond to primary treatments
Despite the low level of published evidence, H2-antihistamines could still be considered as an add on due to their relatively low cost and safety profile but ideally more research would give us a better idea of their efficacy
ORAL STEROIDS
Can consider very infrequent courses of oral prednisolone lasting only a few days for rescue treatment to control severe exacerbations in addition to anti-histamines
SECOND LINE TREATMENT
The algorithm in the BAD (2021) algorithm guidelines state that you can add ‘Omalizumab or Ciclosporin’ alongside a 2nd generation H1-antihistamine if first-line options fail
The European guidelines (EAACI) explicitly states that omalizumab should be tried before ciclosporin because ciclosporin has an inferior adverse effect profile and is not licensed for urticaria
OMALIZUMAB
Omalizumab is a monoclonal antibody that binds to circulating IgE
By binding free IgE it prevents IgE from attaching to the high-affinity FcεRI receptor on mast cells and basophils
It is licensed as an add-on therapy for chronic spontaneous urticaria in patients aged 12 years and older with an inadequate response to H1 antihistamine treatment
Biosimilar availability: An omalizumab biosimilar (Omlyclo) is now available in the UK and has demonstrated comparable efficacy to the originator in phase III CSU trials
Efficacy:
Effective in approximately 85% of patients, with most experiencing rapid symptom relief — often within the first few weeks of treatment.
Across pivotal phase trials, roughly 50–60% of patients achieve mild disease (UAS7 < 6) and 30–40% achieve complete symptom control (UAS7 = 0) on the 300mg dose
Omalizumab in Chronic Spontaneous Urticaria — Pivotal Trials
Omalizumab 300 mg subcutaneously every 4 weeks.
| Trial | Participants | Baseline UAS7 | Primary Endpoint | UAS7 Result at Week 12 |
|---|---|---|---|---|
| ASTERIA I | 319 | ~30.7 | Change in itch severity score at Week 12 | UAS7 ≤ 6: 51.9% vs 11.3% placebo |
| ASTERIA II | 323 | ~30.7 | Change in itch severity score at Week 12 | UAS7 ≤ 6: 65.8% vs 19.0% placebo |
| GLACIAL | 336 | ~31.2 | Safety (efficacy key secondary) | UAS7 ≤ 6: 52.4% vs 12.0% placebo |
| SUNRISE | 136 | ~32.2 | Disease control on Urticaria Control Test | UAS7 ≤ 6: 67.7% (open-label) |
| XTEND-CIU | 206 | ~32.2 | Sustained response and relapse after withdrawal | UAS7 ≤ 6: 58.8% at Week 12 Rose to 73.0% by Week 24; benefit sustained to Week 48 |
Predicting response - the role of endotypes
The two CSU endotypes (type I autoallergic vs type IIb autoimmune) show different response patterns:
Type I autoallergic (high total IgE, negative BAT) — fast responders to omalizumab
Type IIb autoimmune (low IgE, positive BAT, anti-TPO often present) — slow or partial responders to omalizumab; may respond to ciclosporin
Checking total IgE and BAT/BHRA (where available) can therefore help predict who may respond well to omalizumab
UK versus European guideline practical differences
UK prescribing practice around omalizumab is more restrictive than the international (EAACI) guidelines. In the UK, omalizumab is typically given in set 24-week courses after which patients are required to have a treatment break to confirm whether disease is still active — practically this can be tedious for patients who are clearly still symptomatic.
Dose escalation (either increasing to 600mg or shortening the interval to every 2 weeks) is considered off-licence and is not generally permitted — so if the standard 300mg every 4 weeks is not sufficient, the usual next step is to stop omalizumab and switch to ciclosporin.
The EAACI international guidelines are more flexible and explicitly support updosing omalizumab (higher dose, shorter interval, or both) before abandoning it, acknowledging that while this is off-label, patients should be informed and given the chance to respond at a higher dose before moving on to another agent.
Administering omalizumab:
The standard recommended initial dosage is 300 mg administered every 4 weeks.
No routine baseline screening or ongoing blood monitoring is required.
Standard protocol dictates that patients should be monitored under medical supervision for 20 to 30 minutes following their first two to three injections
It is likely that the real-world risk of anaphylaxis in the treatment of CSU is very low
In some cases, what appears to be an adverse reaction may be a small flare-up of urticaria or angioedema immediately following the injection
The risk of anaphylaxis is higher in patients receiving omalizumab for asthma than in those treated for CSU. This may reflect both the more atopic nature of the asthma population and the higher, weight- and IgE-based dosing used in asthma compared with the fixed dose used in CSU
In terms of other side effects, omalizumab is considered very well-tolerated with a strong safety profile, even for long-term use
Common side effects can include headaches and joint pain
Infection risk: It may cause reduced immunity against parasitic infections
CICLOSPORIN
Ciclosporin is used off-label in urticaria
Generally see response rates of ~40–60%, with some patients experience significant symptom relief within 4 weeks
Dosing: Similar as for eczema = 2.5 - 5mg/kg/day, adjusted based on response and tolerability
Duration: 3-6 months course recommended due to risk of cumulative toxicity with prolonged courses
In general, omalizumab is preferred over ciclosporin as the first step when antihistamines have not worked — it has a better safety profile, does not require blood monitoring, and is licensed for this indication.
Ciclosporin is particularly worth considering in patients who have responded poorly to omalizumab and who have features of the type IIb autoimmune endotype — positive BAT/BHRA, low total IgE, basopaenia/eosinopaenia, or anti-TPO positivity.
This endotype tends to be a slow or partial responder to omalizumab but a better responder to ciclosporin
DUPILUMAB
Dupilumab (Dupixent) is a monoclonal antibody that blocks the shared alpha subunit of the IL-4 and IL-13 receptors, thereby inhibiting signalling from both of these type 2 cytokines
It is already widely used in dermatology for atopic dermatitis and nodular prurigo, and has licensed indications in asthma, chronic rhinosinusitis with nasal polyps (CRSwNP) and eosinophilic oesophagitis.
It has now been approved for chronic spontaneous urticaria in the EU, USA and Japan as an add-on to H1 antihistamines.
Mechanism of action in CSU
By blocking IL-4 and IL-13 signalling, dupilumab is thought to work in CSU through several mechanisms including:
Reduced B-cell class-switching to IgE and therefore reduced IgE production over time
Reduced priming and activation of mast cells (as there is less IgE available to bind FcεRI)
Reduced activation of basophils and eosinophils, which also contribute to the inflammatory milieu in CSU
It is not yet known whether other IL-13-targeting agents have similar efficacy in CSU.
Efficacy — the CUPID trials (LIBERTY-CUPID)
The LIBERTY-CUPID programme comprised three phase 3 trials of dupilumab added to a H1 antihistamine in CSU. The primary endpoint was change from baseline in UAS7
| Study | Population | UAS7 change at wk 24 (dupilumab vs placebo) | Outcome |
|---|---|---|---|
| Study A | Omalizumab-naïve | −20.5 vs −12.0 | Met primary endpoint — statistically and clinically significant |
| Study B | Omalizumab-intolerant or inadequate responders | −14.4 vs −8.5 | Did not meet primary endpoint — trend toward benefit only |
| Study C | Omalizumab-naïve (confirmatory) | Met primary endpoint — supported US and EU approval |
Study A
Omalizumab-naïve patients.
Statistically and clinically significant improvement point improvement in UAS7 on dupilumab (-20.5) vs placebo (-12) at week 24
Study B
Patients who were omalizumab-intolerant or had inadequate response to omalizumab.
Showed a trend toward benefit but did not meet its primary endpoint
Effect size was smaller than in Study A: Dupilumab (-14.4) vs placebo (-.8.5)
Study C —
Repeat study in omalizumab-naïve patients which met the primary endpoint
This supported approval in the US and Europe.
When pooled analysis was performed of 289 omalizumab naive patient from the CUPID-A and C study - 43.1% achieved UAS7 ≤ 6 (well controlled) and 30.6% achieved UAS7=0 (urticaria free)
Take home point:
Dupilumab is effective in omalizumab-naïve CSU
Benefit in omalizumab-refractory disease is smaller and less consistent
Consider dupilumab particularly where there is co-existing atopic disease (atopic dermatitis, asthma, CRSwNP).
Extrapolated comparison with omalizumab
Omalizumab remains the most efficacious second-line biologic for CSU on current evidence
Dupilumab can be effective in omalizumab-naïve patients, but benefit in omalizumab-refractory patients appears more modest
Onset of action with dupilumab also appears somewhat slower than with omalizumab or remibrutinib.
Safety
The safety profile in CSU is in keeping with that seen in other indications and is generally very favourable
Conjunctivitis (well recognised in atopic dermatitis) does not appear to have been a significant issue in the urticaria trials.
Where may dupilumab be useful?
Patients with CSU and co-existing type 2 conditions — atopic dermatitis, asthma, CRSwNP, nodular prurigo — where a single agent can target multiple diseases
Patients in whom omalizumab has not been tolerated or is contraindicated, bearing in mind that response in the omalizumab-refractory group is less robust
REMIBRUTINIB
Remibrutinib is an oral, highly selective Bruton's tyrosine kinase (BTK) inhibitor being developed for CSU
Phase 3 trials are complete and approval is anticipated in the first half of 2026
Mechanism of action
BTK is an intracellular signalling molecule expressed in B cells, mast cells, basophils and eosinophils.
In mast cells and basophils, BTK sits downstream of the high-affinity IgE receptor (FcεRI) and is essential for transmitting the activation signal that leads to degranulation. Inhibiting BTK therefore reduces histamine and mediator release.
BTK is also essential for B-cell receptor signalling
Inhibition dampens B-cell activation and autoantibody production — this is relevant in CSU because it targets the upstream generation of the IgG autoantibodies that drive the type IIb endotype
Notably, at CSU therapeutic doses remibrutinib doesn't deplete B cells or meaningfully reduce total IgG/IgM/IgE levels — so immune competence is largely preserved.
A key conceptual point: because BTK acts intracellularly, downstream of FcεRI it is effective in both type I autoallergic (IgE antibodies) and type IIb autoimmune (IgG antibodies) endotypes. This makes it a potential attractive option for patients who are slow or poor responders to omalizumab.
Efficacy — phase 3 data
In the phase 3 REMIX-1 and REMIX-2 trials (Metz et al., NEJM 2025), approximately half of patients treated with remibrutinib 25 mg BD achieved well-controlled disease (UAS7 ≤ 6) at week 12 — 49.8% in REMIX-1 and 46.8% in REMIX-2, compared with 24.8% and 19.6% on placebo (both p < 0.001). This response was sustained through week 24
Rapid onset of action was seen with separation from placebo evident as early as week one
Both trials included a 28-week open-label extension in which all patients — including those originally randomised to placebo — received remibrutinib 25 mg BD to week 52, with efficacy sustained throughout and nearly half of treated patients free of itch and hives (UAS7 = 0) by the end of the study.
Approximately one-third of patients in the pooled REMIX population had prior exposure to anti-IgE biologic therapy, and a pre-specified subgroup analysis (Mosnaim et al., JACI 2024) showed remibrutinib was similarly effective in omalizumab-experienced and omalizumab-naïve patients, with UAS7 = 0 achieved in 32.1% versus 28.4% at week 12, supporting its role as an option in omalizumab non-responders.
Comparison with omalizumab
On current cross-trial comparison, omalizumab probably remains the most efficacious second-line agent overall, but remibrutinib appears to be at least as fast-acting as omalizumab and faster than dupilumab. Its oral route is a significant practical advantage.
Safety
Only around one year of safety data is currently available, so long-term safety remains to be established. Issues reported in trials include:
Transient liver enzyme elevations (generally not exceeding three times the upper limit of normal)
Mildly increased rate of upper respiratory tract infections. Total IgG, IgM and IgE levels appear stable, but whether vaccine responses are preserved is not yet fully characterised
Petechiae — reported approximately three times more commonly than placebo, but without clinically significant bleeding issues
Where remibrutinib may be particularly useful
Patients who prefer (or would benefit from) an oral therapy over injectable biologics
Patients with type IIb autoimmune endotype features (positive BAT/BHRA, low total IgE, anti-TPO, basopaenia/eosinopaenia) who are likely to be slow or poor responders to omalizumab — as remibrutinib acts downstream of IgE
Patients who have failed omalizumab
Note: BTK is the same molecule that is defective in X-linked (Bruton) agammaglobulinaemia, and BTK inhibitors are already approved for haematological malignancies including CLL, mantle cell lymphoma, marginal zone lymphoma and Waldenström's macroglobulinaemia
Summary of pivotal trials
| Timepoint | Complete response (UAS7 = 0) |
Partial response (UAS7 ≤ 6) |
Placebo (UAS7 = 0) |
Placebo (UAS7 ≤ 6) |
Reference trials |
|---|---|---|---|---|---|
| Omalizumab | |||||
| Week 12 | ~34–44% | ~52–66% | ~5–10% | ~11–20% | ASTERIA I / II, GLACIAL |
| Week 24 | ~43–48% | ~56–62% | ~4–13% | ~17–25% | |
| Dupilumab | |||||
| Week 24 | ~13–31% | ~24–46% | ~9–18% | ~19–24% | LIBERTY-CSU CUPID A, B, C |
| Remibrutinib | |||||
| Week 12 | ~28–31% | ~47–50% | ~7–11% | ~20–25% | REMIX-1, REMIX-2 |
| Week 24 | ~36% | ~52–55% | ~16–20% | ~28–35% | |
Updated treatment algorithm
New international guidelines (publication pending at time of writing) are expected to position omalizumab, dupilumab and remibrutinib as alternative second-line options following uptitrated H1 antihistamines, with choice guided by patient preference, comorbidities and endotype where assessable
Ciclosporin moves further down the algorithm.
THIRD LINE TREATMENT
With the arrival of omalizumab (and its biosimilar omlyclo), dupilumab and the imminent approval of remibrutinib, third-line agents are now rarely needed in practice
The evidence base for these agents is weak - largely case series and small uncontrolled studies - and most have moved into a "historical" tier in the international guidelines
If a patient has failed uptitrated antihistamines, omalizumab (including uptitration where permitted), dupilumab/remibrutinib where available, and ciclosporin, then options to consider (in no particular order) include:
- Methotrexate
- Azathioprine
- Mycophenolate mofetil
- Dapsone (may have a role in delayed pressure urticaria)
- Narrowband UVB (may have a role in symptomatic dermographism)
- Hydroxychloroquine
- Doxepin (tricyclic with potent H1 antihistamine activity)
- IVIG
- Tranexamic acid (if angioedema is the predominant feature)
At this stage it is worth revisiting the diagnosis — ensure urticarial vasculitis, autoinflammatory syndromes and mast cell disorders have been excluded
SPECIFIC TREATMENT INDUCIBLE URTICARIAS
General principles
The general approach mirrors that of CSU:
Avoid the trigger where possible (not always practical - eg friction in symptomatic dermographism)
Start with a 2nd generation H1 antihistamine, uptitrated up to fourfold if needed
If antihistamines fail, omalizumab is the next step and is now the best-evidenced second-line option across the inducible urticarias
Omalizumab in CIndU
Although omalizumab is only formally licensed for CSU, there is good evidence supporting its use off-label in CIndU, particularly for the three most common subtypes:
Symptomatic dermographism - supported by a randomised placebo-controlled trial
Cold urticaria - supported by a randomised placebo-controlled trial
Solar urticaria - supported by case series and open-label studies
Evidence is more limited but generally supportive for cholinergic, delayed pressure and heat contact urticarias, and largely anecdotal for aquagenic, vibratory and contact urticarias
In real-world cohorts roughly 70% of CIndU patients achieve a complete or good response to omalizumab, which is broadly comparable to its efficacy in CSU
Reference: Soegiharto R, et al. Omalizumab is effective and safe in chronic inducible urticaria (CIndU): Real-world data from a large multi-national UCARE study. Allergy. 2025.
Subtype-specific considerations
Although the overall approach is similar, a few subtypes have additional or alternative treatment options worth knowing:
Cholinergic urticaria - consider anticholinergics (eg oxybutynin), beta blockers (eg propranolol), or danazol if antihistamines and omalizumab fail.
Response to omalizumab is less predictable than in dermographism or cold
urticaria
Cold urticaria - patients should carry an adrenaline auto-injector given the risk of anaphylaxis. Ciclosporin can be considered as an alternative in refractory cases
Delayed pressure urticaria - often the hardest CIndU to treat. Dapsone or sulfasalazine can be considered. Montelukast has some supporting evidence here
Solar urticaria - following photoinvestigation to identify the action spectrum, prophylactic phototherapy (hardening using the relevant wavelength) can be very
effective alongside sun avoidance and protection
Symptomatic dermographism - narrowband UVB (and failing that PUVA) can be useful as an adjunct
Aquagenic urticaria - barrier creams (eg petrolatum) applied before water exposure can be helpful
Future directions
Barzolvolimab, an anti-KIT monoclonal antibody that depletes
mast cells, has shown striking efficacy in early trials for
symptomatic dermographism and cold urticaria and is a
treatment to watch in this space
ANGIOEDEMA WITHOUT WEALS
Angioedema occurring without weals raises a different differential to the urticarial picture, because mediators other than histamine come into play. The main causes are:
Idiopathic
Drug induced (eg ACE inhibitors, NSAIDs, antibiotics)
C1 esterase inhibitor deficiency (hereditary or acquired)
Hereditary angioedema with normal C1 esterase inhibitor
The bradykinin pathway:
Bradykinin is a vasoactive peptide normally produced at sites of tissue injury. Its job is to cause vasodilation and increased vascular permeability, allowing plasma proteins and immune cells to reach damaged tissue as part of the acute inflammatory response. It is generated through a plasma protein cascade known as the kinin–kallikrein system.
In simple terms:
Factor XII is activated to Factor XIIa on contact with damaged endothelium
Factor XIIa converts prekallikrein into kallikrein
Kallikrein cleaves high molecular weight kininogen to release bradykinin
Bradykinin binds the B2 receptor on vascular endothelium, causing vasodilation and plasma leakage
Two brakes normally keep this loop in check:
C1 inhibitor inhibits Factor XIIa and kallikrein, shutting down bradykinin production
Angiotensin converting enzyme (ACE) degrades bradykinin once it has been produced
In angioedema, bradykinin accumulates either because it is being overproduced (as in C1 inhibitor deficiency, where the brake on kallikrein is lost) or because it is not being broken down (as with ACE inhibitor use, where the enzyme that degrades bradykinin is blocked)
Because this pathway is entirely independent of histamine, bradykinin-mediated angioedema does not respond to antihistamines, adrenaline or corticosteroids
Clinical clues
Most cases of angioedema without weals are idiopathic, but it is important not to miss drug-induced angioedema (eg ACEi, NSAIDs, antibiotics) or C1 esterase inhibitor deficiency, as both can cause life-threatening airway swelling and neither responds to standard angioedema treatment.
Angioedema of the gastrointestinal tract is common in C1 esterase inhibitor deficiency, so always ask about recurrent abdominal pain.
Hereditary angioedema:
Hereditary angioedema (HAE) is traditionally classified into Types I, II and III, and these terms are still widely used.
Note: Current 2025 WAO/EAACI nomenclature unifies Types I and II under HAE-C1-INH (C1 inhibitor deficiency vs dysfunction), and refers to the older "Type III" as HAE with normal C1 inhibitor (HAE-nC1-INH), with subtypes named by their causative mutation
Type I and II are autosomal dominant and due to mutations in the SERPING1 gene, which encodes C1 inhibitor:
Type I (~85%): C1 esterase inhibitor levels decreased
Type II (~15%): C1 esterase inhibitor levels normal or increased, but functional levels decreased
Type III (HAE with normal C1 inhibitor) is much rarer. C4 and C1 esterase inhibitor levels are typically normal. Causative mutations include F12 (Factor XII), PLG (plasminogen), KNG1 (kininogen) and ANGPT1 (angiopoietin-1), among others. It is more common in females and can be aggravated by oestrogens.
Acquired angioedema:
Acquired angioedema is due to depletion or destruction of C1 esterase inhibitor through immune complexes or autoantibodies. It is divided into:
Type I: associated with lymphoproliferative disorders (B-cell lymphoma, NHL, multiple myeloma)
Type II: associated with autoimmune disease, with autoantibodies against C1 inhibitor
Both types show low C1 inhibitor level and function, low C4, and — importantly — low C1q. The reduced C1q level is what differentiates acquired from hereditary angioedema, where C1q is normal
Investigations:
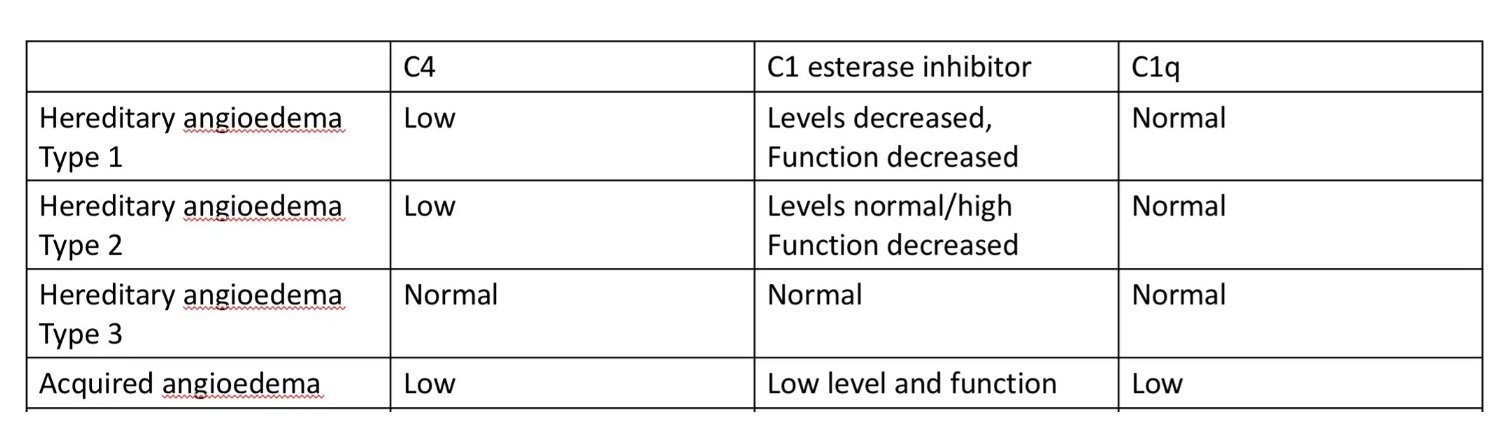
Practical tips:
C4 is low both between and during attacks in C1 esterase inhibitor deficiency, making it a useful screening test
If C4 is low, check C1 inhibitor level and function
Check C1q if acquired angioedema is suspected
HAE Type III often requires genetic testing to confirm the diagnosis
Refer all forms of C1 esterase inhibitor deficiency to immunology.
Treatment:
Antihistamines, adrenaline and corticosteroids are largely ineffective in bradykinin-mediated angioedema. Treatment is built around three goals: treating acute attacks, providing cover before known triggers, and reducing attack frequency.
Acute attacks (on-demand):
C1 esterase inhibitor concentrate (plasma-derived or recombinant)
Icatibant — a B2 receptor antagonist given subcutaneously, which patients can self-administer
Fresh frozen plasma is now reserved as a fallback only when specific therapies are unavailable
Short-term prophylaxis (eg before dental work or surgery):
C1 esterase inhibitor concentrate is preferred
Long-term prophylaxis:
Modern first-line options are the kallikrein inhibitors lanadelumab (subcutaneous monoclonal antibody) and berotralstat (oral, once daily)
Newer agents include donidalorsen (reduces prekallikrein production) and garadacimab (Factor XIIa inhibitor given monthly subcutaneously)
Attenuated androgens (danazol, stanozolol) and tranexamic acid are now considered historical options, used only where modern targeted therapies are unavailable
For acquired angioedema, much higher doses of C1 inhibitor concentrate are typically required, and the underlying lymphoproliferative or autoimmune driver should be sought and treated.
URTICARIAL VASCULITIS
Urticarial vasculitis (UV) is a rare small-vessel leukocytoclastic vasculitis that presents with urticarial wheals but differs from chronic spontaneous urticaria in some important ways
Typical patients are middle-aged women, and it's easily mistaken for CSU at first glance
Clinical clues to UV rather than CSU:
Individual lesions last more than 24 hours (in contrast to CSU, where wheals come and go within 24 hours)
Lesions are often painful or burning rather than purely itchy
Lesions resolve leaving residual petechiae, purpura, or post-inflammatory hyperpigmentation/bruising
Angioedema occurs in around 50% of UV patients
Systemic features — fever, arthralgia, malaise, abdominal pain, or eye inflammation
Raised inflammatory markers (CRP, ESR) — often out of keeping with what you'd expect from CSU
Dipstick may show blood or protein in the urine
Pathogenesis:
UV is driven by immune complex deposition in small vessel walls, with complement activation and neutrophil recruitment causing leukocytoclastic damage to the vessels themselves. This is what distinguishes it from CSU — in UV you get true vessel wall damage with red cell extravasation, rather than just transient oedema from mast cell degranulation.
Normocomplentaemic UV (NUV)
This is the commonest form, accounting for roughly 70–80% of cases
Complement levels (C3, C4, C1q) are normal
Disease tends to be skin-limited or associated with only mild systemic features such as arthralgia, low-grade fever, or malaise
Prognosis is generally favourable, and the condition is often self-limiting over months to a couple of years
Associations with underlying connective tissue disease are much weaker than hypocomplementaemic UV
Most cases are idiopathic but it can be associated with a range of drugs/conditions
NUV: most cases idiopathic — recognised associations below
Drugs
- ACE inhibitors
- NSAIDs
- Thiazide diuretics
- Penicillins
- Fluoxetine
- (others reported)
Infections
- Hepatitis B
- Hepatitis C
- Infectious mononucleosis
- Lyme disease
Paraproteinaemia
- IgM gammopathy
Connective tissue disease
Much more typical of HUV
Occasional NUV cases evolve into or coexist with connective tissue disease
Malignancy
- Haematological (more common)
- Solid organ (less common)
Hypocomplementaemic UV (HUV)
Hypocomplementaemic urticarial vasculitis (HUV) sits in the middle of the urticarial vasculitis spectrum — more clinically significant than NUV but without meeting the full criteria for HUVS
The defining feature is persistent low C3 and/or low C4, reflecting ongoing immune complex-mediated complement consumption
Clinically the skin lesions are similar to NUV but systemic involvement is far more common
Arthralgia and arthritis occur in the majority of patients, and angio-oedema, fever, and malaise are frequent
Extracutaneous organ involvement (pulmonary, ocular, renal, gastrointestinal) is less universal than in HUVS, but a proportion of patients will progress to meet HUVS criteria over time.
Characterised by low C3 and/or C4, with C1q often also reduced. Disease is more likely to be systemic, with arthralgia, fever, abdominal pain, pulmonary and renal involvement.
Hypocomplementaemic urticarial vasculitis
Low C3 and/or C4 — stronger autoimmune weighting than NUV
Connective tissue disease
- SLE
- Sjögren syndrome
- Mixed connective tissue disease
May precede, coincide with, or emerge during SLE.
Less common
Drugs
ACE inhibitors, NSAIDs, thiazide diuretics, penicillins.
Infections
Hepatitis B and C, EBV / infectious mononucleosis, Lyme disease.
Around 50% of HUV patients will either have or go on to develop an underlying CTD, so these patients need long-term follow-up and a low threshold for repeat autoimmune serology over time
Hypocomplementaemic urticarial vasculitis syndrome (HUVS)
HUVS is the severe end of the urticarial vasculitis spectrum and is defined by strict criteria (Schwartz criteria):
Schwartz criteria for HUVS
Both major criteria plus at least two minor criteria required
Major criteria
Both required
- Recurrent urticarial lesions for more than 6 months
- Hypocomplementaemia
Minor criteria
At least two required
- Leukocytoclastic vasculitis on biopsy
- Arthralgia or arthritis
- Glomerulonephritis
- Uveitis or episcleritis
- Recurrent abdominal pain
- Positive anti-C1q antibodies with low C1q
The hallmark of HUVS is the presence of anti-C1q autoantibodies and markedly reduced C1q — both are relatively specific and help distinguish HUVS from HUV
There is substantial clinical and serological overlap with SLE, and a significant proportion of HUVS patients meet SLE classification criteria either at presentation or over time
Organ involvement in HUVS is multi-system:
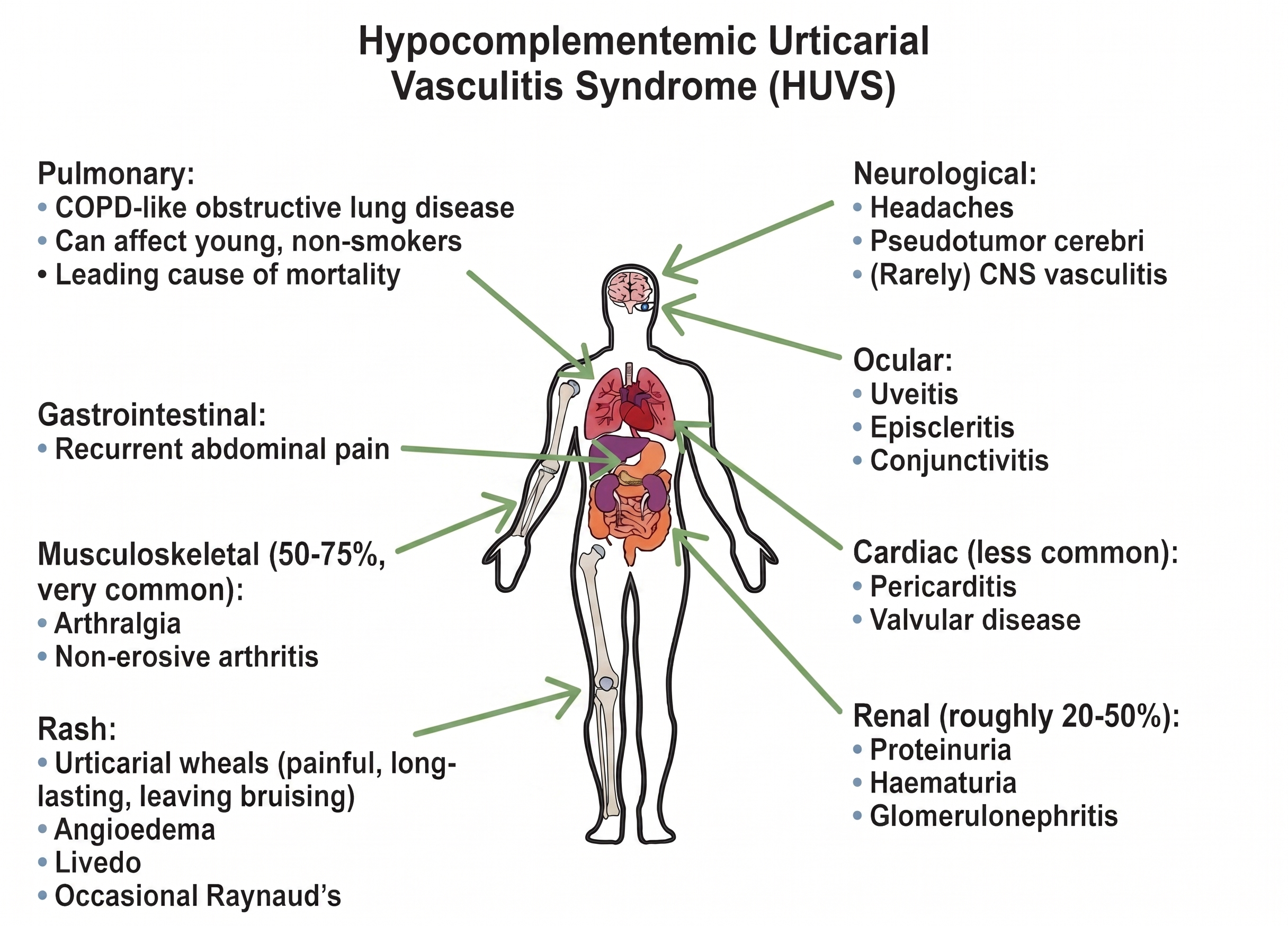
HUVS needs a proactive, systems-based assessment at diagnosis — urinalysis, pulmonary function tests, and ophthalmology review — with ongoing multidisciplinary follow-up including rheumatology and respiratory.
Investigations:
If considering Urticarial Vascuilitis a workup could include:
Skin biopsy — essential for diagnosis. Ideally biopsy a lesion that's been present for less than 48 hours
FBC, CRP, ESR — inflammatory markers often raised
Complement levels — C3, C4, and C1q (low C1q suggests HUVS)
ANA, ENA, anti-dsDNA — to look for SLE or other CTD
Anti-C1q antibodies — if HUVS suspected
U&E, urinalysis — to screen for renal involvement
LFTs, hepatitis B and C serology
Serum and urine protein electrophoresis — to exclude a paraprotein
Consider extended workup guided by any systemic features (eg CXR and PFTs is suspect HUVS with respiratory involvement)
Histology:
A skin biopsy is required to confirm the diagnosis, though findings can be subtle and aren't always dramatic. Look for:
Leukocytoclasia (nuclear dust from fragmenting neutrophils)
Fibrinoid necrosis of vessel walls (classical but not always seen)
Endothelial swelling and damage
Red cell extravasation
Perivascular neutrophilic infiltrate
Management:
Mild skin-limited disease
Patient is well, no systemic symptoms, normal complement, lesions manageable.
Non-sedating H1 antihistamines (cetirizine, fexofenadine, bilastine) up-dosed to 4x standard dose, as per chronic urticaria guidelines. Often only partially effective
Potent topical corticosteroids for individual lesions.
NSAIDs (e.g. indomethacin) for arthralgia and lesional discomfort — useful adjunct if no contraindications
Identify and remove triggers
Moderate - Persistent skin disease with mild systemic features
Frequent lesions, arthralgia, fatigue, but no organ-threatening disease.
Colchicine 500 micrograms BD–TDS — well-tolerated first-line systemic agent. Watch for GI side effects and check renal function
Dapsone 50–100 mg daily — effective for neutrophil-rich vasculitis. Check G6PD, baseline FBC and LFTs, monitor for haemolysis and methaemoglobinaemia
Hydroxychloroquine 200–400 mg daily — particularly useful in HUV and when there is overlap with connective tissue disease
Short prednisolone courses (e.g. 20–30 mg tapering over 4–6 weeks) for flares, but avoid as maintenance
Severe - HUV, organ involvement or refractory disease
Renal, pulmonary, CNS, or ocular involvement, or skin disease refractory to the above.
Multidisciplinary input: rheumatology, nephrology, respiratory as required.
Oral corticosteroids (prednisolone 0.5–1 mg/kg/day) to induce remission, with a steroid-sparing agent started in parallel.
Immunosuppressants: mycophenolate mofetil, azathioprine, or methotrexate
Rituximab — increasing evidence in refractory HUV and HUVS, particularly with pulmonary involvement
Omalizumab — case reports and small series suggest benefit in NUV behaving like chronic urticaria; less convincing in HUV.
IVIG or cyclophosphamide reserved for life- or organ-threatening disease, in conjunction with rheumatology.
AUTOINFLAMMATORY SYNDROMES
Pathophysiology of Autoinflammatory Urticaria
Autoinflammatory syndromes are characterized by primary dysfunction of the innate immune system. Unlike autoimmune diseases, they lack autoantibodies or antigen-specific T-cell responses
The underlying mechanisms vary. Some, such as CAPS, are monogenic disorders caused by inherited gain-of-function mutations in inflammasome components. Others, such as Schnitzler syndrome and adult-onset Still's disease, are acquired and multifactorial, with disease activity driven by environmental triggers or dysregulated proteins such as a monoclonal paraprotein
Regardless of the initial trigger, the common endpoint is the same: inflammasome hyperactivation (such as the NLRP3 inflammasome) leading to a massive, uninhibited overproduction of pro-inflammatory cytokines, particularly Interleukin-1β (IL-1β)
Clinically, the "urticarial" rashes seen in these syndromes represent a neutrophilic urticarial dermatosis rather than classical histamine-driven mast cell degranulation
They differ from typical spontaneous urticaria in several key ways:
Lesions frequently cause burning or pain rather than pruritus
Often persist for >24 hours in a fixed location
May leave behind residual purpura or hyperpigmentation
Are notoriously refractory to standard antihistamine therapy
Key Urticarial Autoinflammatory Syndromes
Schnitzler’s Syndrome
A rare, late-onset (acquired) autoinflammatory disorder
Diagnosis typically requires the presence of an urticarial rash and a monoclonal protein, alongside minor clinical criteria
Rash: Chronic, non-pruritic (or mildly pruritic) urticarial eruption
Defining Feature: Monoclonal IgM gammopathy (essential for diagnosis; rarely IgG)
Systemic Symptoms: Recurrent fevers, severe bone and joint pain, lymphadenopathy, and hepatosplenomegaly
Labs: Markedly elevated acute phase reactants (CRP/ESR) and leukocytosis
Cryopyrin-Associated Periodic Syndromes (CAPS)
CAPS represents a spectrum of rare, autosomal dominant autoinflammatory diseases caused by gain-of-function mutations in the NLRP3 gene. This leads to continuous cryopyrin-inflammasome assembly and uninhibited IL-1β release. Patients present with a non-pruritic, neutrophilic urticarial rash and systemic inflammation.
The CAPS spectrum ranges from mild to severe:
Familial Cold Autoinflammatory Syndrome (FCAS): The mildest form of CAPS.
Features: Episodes of an urticaria-like rash, low-grade fevers, conjunctivitis, and severe arthralgia strictly triggered by generalized exposure to cold temperatures (typically within 1–2 hours of exposure). It usually presents in infancy or early childhood.
Muckle-Wells Syndrome (MWS): An intermediate severity form of CAPS.
Features: Spontaneous, episodic flares of urticarial rash, fevers, and joint pain that are not strictly dependent on cold exposure.
Complications: MWS is distinctly complicated by progressive sensorineural hearing loss (typically starting in adolescence) and a high risk of secondary AA amyloidosis, which can lead to renal failure.
Neonatal-onset Multisystem Inflammatory Disease (NOMID)
This is the most severe form of CAPS
Characterised by continuous severe inflammation, severe arthropathy, and central nervous system involvement
Adult-Onset Still’s Disease (AOSD)
A rare, polygenic systemic autoinflammatory disorder of unknown etiology, characterized by a classic clinical triad.
Rash: An evanescent, salmon-pink maculopapular rash that is typically non-pruritic. It characteristically flares in tandem with fever spikes and often appears on the trunk or extremities. (While classic AOSD features this salmon-pink rash, persistent atypical urticarial lesions are also increasingly recognized).
Systemic Symptoms: Daily high-spiking (quotidian) fevers and severe polyarthralgia/arthritis.
Labs: Extreme hyperferritinemia (often >1000 ng/mL, with a low proportion of glycosylated ferritin), prominent neutrophilic leukocytosis, and elevated liver transaminases.
Targeted Management
Because the inflammation and cutaneous manifestations in these syndromes are heavily mediated by IL-1 overproduction, standard therapies (such as H1/H2 antihistamines, corticosteroids, or omalizumab) generally yield poor responses.
These conditions typically exhibit a dramatic and rapid response to targeted IL-1 blockade. The mainstays of treatment include:
Anakinra: A recombinant, non-glycosylated form of the human IL-1 receptor antagonist (IL-1Ra) with a short half-life, administered via daily subcutaneous injection.
Canakinumab: A human monoclonal antibody that specifically neutralizes IL-1β, administered subcutaneously every 4 to 8 weeks.
CONTACT URTICARIA
Contact urticaria is a wheal-and-flare reaction occurring within minutes of skin or mucosal contact with a triggering substance, usually settling within an hour. The trigger is direct external contact rather than a spontaneous or physical stimulus.
Two mechanisms are recognised:
Non-immunological — the commonest form. No prior sensitisation needed. Classic culprits are nettles, and preservatives and fragrances in cosmetics and foods (benzoic acid, cinnamic aldehyde, sorbic acid).
Immunological — IgE-mediated and requires prior sensitisation. The classic trigger is natural rubber latex; foods (fish, shellfish, raw fruit and vegetables) and animal proteins are also recognised. These patients are at risk of anaphylaxis.
Clinical points:
Suspect in any patient with immediate wheals at sites of contact — hands in occupational cases, perioral with foods
Take an occupational and exposure history
Management is trigger avoidance, antihistamines for symptoms, and an adrenaline auto-injector for anyone with a history of systemic reactions
Latex-allergic patients can also react to certain fruits — classically banana, avocado, kiwi and chestnut — due to cross-reactivity between latex and plant defence proteins (the "latex–fruit syndrome"). This can present as oral allergy syndrome, contact urticaria or in a minority - generalised urticaria, angioedema and even anaphylaxis if they ingest large amounts of the food